





Abstract
Motivation: Human pluripotent stem cell lines persist in culture as a heterogeneous population of SSEA3 positive and SSEA3 negative cells. Tracking individual stem cells in real time can elucidate the kinetics of cells switching between the SSEA3 positive and negative substates. However, identifying a cell's substate at all time points within a cell lineage tree is technically difficult.
Results: A variational Bayesian Expectation Maximization (EM) with smoothed probabilities (VBEMS) algorithm for hidden Markov trees (HMT) is proposed for incomplete tree structured data. The full posterior of the HMT parameters is determined and the underflow problems associated with previous algorithms are eliminated. Example results for the prediction of the types of cells in synthetic and real stem cell lineage trees are presented.
Availability:The Matlab code for the VBEMS algorithm is freely available at http://www.acse.dept.shef.ac.uk/repository/vbems_lineage_tree/VBEMS.ZIP
Contact: [email protected]
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
A central problem in stem cell biology is how a range of specialized cell types arises from a pool of stem cell progenitors. It has been reported that populations of seemingly homogeneous stem cells consist of cells which fluctuate between interchangeable sub-states (Chambers et al., 2007; Chang et al., 2008; Hayashi et al., 2008). These substates represent functionally distinct cells, which respond to stimuli in a dissimilar manner, generating diverse differentiated progeny. Heterogeneity within stem cell cultures is easily overlooked as many commonly used investigative methods rely on pooling of material before analysis. Tracking the fate of individual cells, provides a powerful method to interrogate cells in their undifferentiated state and during their differentiation. At present, the technology of tracking and monitoring cellular division and cell fate relies on the use of cells modified to carry fluorescent reporter elements to monitor changes in expression of genes known to be associated with particular cellular fates. The use of fluorescent reporter technologies is potentially limiting as it inevitably involves the alteration of the cells and realistically can only be employed to monitor up to three different parameters simultaneously. Labelling of cells using specific antibodies to cell surface proteins allows for limited monitoring of phenotype in real time, but due to the turnover of antigens in live cells requires repeated relabelling of cells with the potential perturbation of the cells characteristics. One solution to these problems is to utilize probabilistic techniques of analysis to reconstruct cell lineage trees based on observations gathered in real time (cells division rate) and combine this with particular surface antigen expression (i.e. phenotypic) information gathered at the end of the set of cell divisions being monitored.
Human embryonic stem (hES) cells spontaneously differentiate and must be constantly monitored for signs of overt differentiation. The cell surface antigen SSEA3 is rapidly down-regulated as hES cells differentiate to more mature cell types (Draper et al., 2002) and thus can serve as a useful readout of the changes in a cell's state from undifferentiated to differentiated. However, hES cells grow in tight colonies and are not amenable to real-time tracking of individual cells; therefore, the NTERA2 cell line has been used as a surrogate. The NTERA2 pluripotent cell line consists of stem cells that express the SSEA3 surface antigen at varying levels. SSEA3 expression levels have functional implications as its expression positively correlates with the probability of a NTERA2 cell to clonally expand (Andrews et al., 1984). Notably, it has been observed that NTERA2 stem cells interconvert between a SSEA3 positive and SSEA3 negative state. To determine the kinetics of this interconversion, SSEA3 negative NTERA2 cells have been obtained by fluorescent activated cell sorting and monitored by time-lapse analysis. In this study, the reconstruction of the lineage trees is based on the observations of the SSEA3 expression of the cells, where the SSEA3 expression of the cells is known only at the root and the leaf levels. There are three potential discrete states in the lineage trees: SSEA3Positive, SSEA3Negative or dead. The challenge consists of determining the SSEA3 expression of each cell at all levels within the lineage trees. In order to achieve this, we propose a variational Bayesian (VB) with smoothed probabilities implementation of hidden Markov trees (HMTs) which represents an analysing tool for any incomplete tree structured data. We take as our starting point the hidden Markov models (HMM), which are used in various fields such as speech recognition, financial time series prediction (Bulla and Bulla, 2006), natural language processing, ion channel kinetics (Fredkin and Rice, 1997) and general data compression (Lee, 2002). They have played important roles in the modelling and analysis of biological sequences, in particular DNA (Beerenwinkel and Drton, 2007; Schliep et al., 2005) and they have proven to be useful tools for statistical signal and image processing.
Baum and colleagues (1970) developed the core theory of HMMs. In 1972, they proposed the forward–backward algorithm as an iterative technique for the maximum likelihood (ML) statistical estimation of probabilistic functions of Markov chains. Devijver (1985) demonstrated that the computation of joint likelihoods in Baum's algorithm could be converted to the computation of posterior probabilities. The resulting algorithm was similar to Baum's except for the presence of a scaling factor suggested by Levinson et al. (1983) which was robust to computational underflow. Further developments in HMMs have come from the work of Mackay (1997), Beal and Ghahramani (2003), Watanabe et al. (2004) and Ji et al. (2006) in which they applied a VB approach to these models.
HMT models have been proposed by Crouse et al. (1997) for modelling the statistical dependencies between wavelet coefficients in signal processing. They have been applied successfully to image de-noising and segmentation (Bharadwaj and Carin, 2002; Choi and Baraniuk, 2001; Romberg et al., 2001) to signal processing and classification (Diligenti et al., 2003; Durand et al., 2004) and to tree structured data modelling (Beerenwinkel and Drton, 2007; Durand et al., 2005). The forward–backward algorithm proposed by Baum was transposed to the HMTs context by Crouse et al. (1997). The resulting algorithm has been called the upward–downward algorithm, but it suffered computational underflow problems as in Baum's algorithm. The upward–downward recursions have been proposed by Ronen et al. (1995) for the E-step in ML estimation of dependence tree models with missing observations. The upward–downward algorithm was revisited by Durand et al. (2004) who made changes to solve the computational underflow problems by adapting the ideas from Devijver's changes to the forward–backward algorithm. Romberg et al. (2001) proposed a Bayesian HMT model for image processing using wavelets and later, Dasgupta and Carin (2006) developed the VB-HMT model based on the model proposed by Crouse et al. with a similar application.
In this study, we derive the VB with smoothed probabilities implementation of HMTs. We extend Durand's HMT framework (Durand et al., 2004) to VB with the critical embodiment of prior probability distributions. Inclusion of prior probability distributions of a class of HMT models such as in the case of cell lineages is essential to avoid ill-posedness of the estimation problem. We demonstrate this through an application to modelling stem cell lineages in simulation and using real data.
2 HMT MODEL
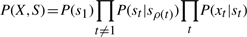
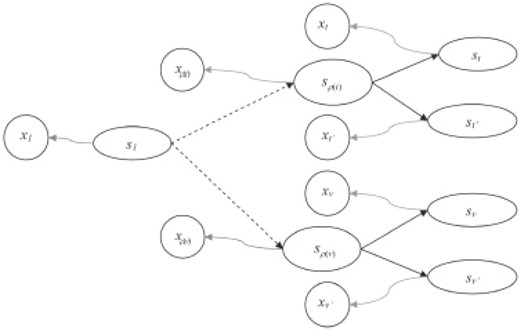
HMT representation with observed nodes (x) and hidden nodes (s). The straight arrows show the state transitions, curly arrows show emission probabilities, dashed arrows show state transitions with possibility of existence of intermediate nodes.

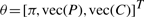
3 ML ESTIMATION FOR HMT MODEL

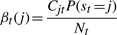
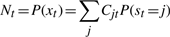
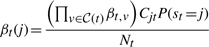
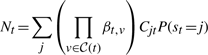
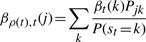
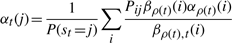
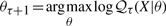
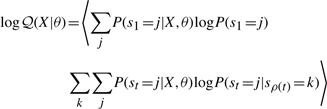
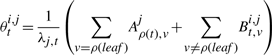
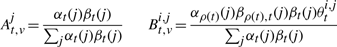
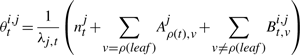
The second upwards–downward algorithm mentioned above represents the base of the E-step of the VB-EM algorithm with smoothed probabilities developed in the next section.
4 VB-EM WITH SMOOTHED PROBABILITIES ALGORITHM
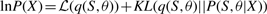
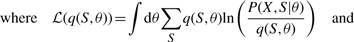
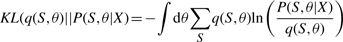
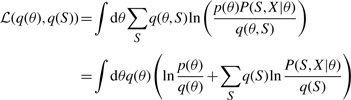
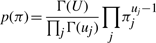
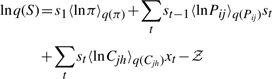
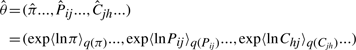
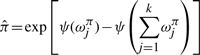
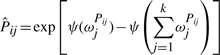
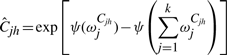
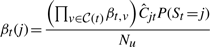
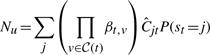
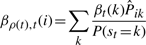
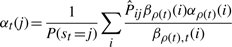
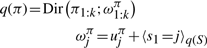
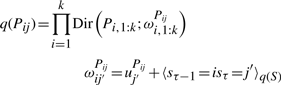
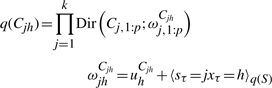
The variational posterior distribution has the same functional form of the Dirichlet distribution. The hyperparameters are equal to the sum between the strength of the prior distribution and statistics of the hidden state and observations which are functions of α and β determined at the E-step.
5 MATERIALS AND BIOLOGICAL METHODS.
Live NTERA2 cells were stained as a cell suspension for SSEA3 expression and SSEA3 positive cells were isolated by fluorescence activated cell sorting. Sorted cells were plated at 15 000 cells/cm2 and placed inside a preheated (37‰) humidified chamber containing 5% CO2. Images of cells were captured every 10 min with the use of an automated motorized stage (Prior) using an inverted Olympus microscope IX70 connected to a hamamatsu camera. After 100 h the cells were fixed with 4% Pfa and stained for SSEA3 expression. The antibody stained cells were then returned to the Olympus IX70 and immunofluorescent images captured at the identical fields of view used for obtaining phase contrast time-lapse images. Phase contrast images from each field of view were compiled to generate cinematic movies that comprise of frames, each representing 10 min intervals. The movies were then replayed and a lineage tree generated for each original cell, with every cell division and cell death event noted relative to time. Starting cells which failed to survive beyond the initial 24 h were excluded from analysis. After referring to the relevant immunofluorescent image, a phenotype (SSEA3−/SSEA3+) could be assigned to each cell of the lineage trees.
6 DISCUSSION AND RESULTS
In this study, we address the problem of how cells with particular characteristics arise from common stem cell progenitors. We utilize probabilistic techniques of analysis to reconstruct the incomplete cell lineage trees obtained from tracking and monitoring the cellular division and fate. The necessity of stochastic modelling techniques comes from the limitations of the available technology for monitoring the stem cells phenotype, in real time. The monitoring techniques involve the use of fluorescent reporter technologies which alter the cells. The experimental lineage trees used here resulted from the combination of the observations gathered in real time on the NTERA2 cells division rate and the SSEA3 marker antigen expression information summoned at the end of the set of cell divisions. The topology of the trees along with the SSEA3 expression of the cells at the root and leaf levels are observed. The potential discrete states of the stem cells in the lineage trees are: SSEA3Positive, SSEA3Negative or dead.
We proposed a HMT model of which parameters were estimated using the VB approach with smoothed probabilities. The VBS-HMT model is applied to the incomplete stem cells lineage tree data. The experimental data used in this study consists of 30 lineage trees, where just the type of cells at the start and at the end of each tree is known. The model developed here confronts the challenge of stem cell lineage tree reconstruction by determining the most likely state tree corresponding to the observed stem cell lineage tree.
Based only on partial observed experimental stem cell lineage tree data, it is challenging to validate the model proposed here and to compare it with other models from the literature. Hence, we created artificial cell lineage trees which give us much more information and have a similar topology to experimental lineage trees. The use of synthetic data enables us to compare four different models: VBS-HMT, ML-based HMT (ML-HMT), ML with prior information-based HMT (ML+P-HMT) and the classic VB-HMT. The performance is evaluated based on the accuracy of prediction of the state of cells within the trees and on the estimation of the parameters.
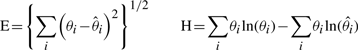
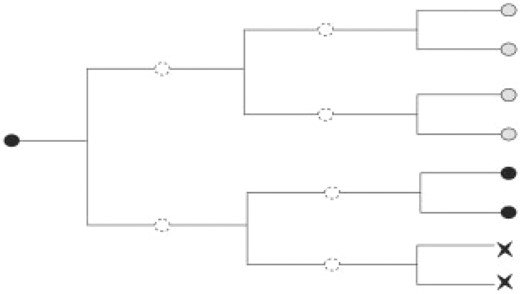
Diagram representing artificial cell lineage tree where light grey cells are positive definite, black cells are negative definite, the X shape cells are dead cells and the white ones, with dashed contour, are unobservable.
Without any supplementary prior information the VB approach proved to be superior to the ML method. The ML approach obtains a single estimate of the HMT model parameter, whereas the VB-HMT obtains a posterior distribution for the HMT model parameters. The VBS-HMT model performed better than the VB-HMT model based on the original upward–downward algorithm. As shown in Table 1, the estimated values of parameters θVBS−HMT is closer to the values of true parameter θ compared with θML+P or θVB−HMT, i.e. the error and relative entropy for the VBS–HMT model are smaller.
Parameter estimation performance of the models applied to the tree structured data
| Model | E | H |
|---|---|---|
| ML-HMT | 0.0218 | 0.1267 |
| ML+P-HMT | 0.0037 | 0.0876 |
| VB-HMT | 0.0062 | 0.0963 |
| VBS-HMT | 0.0012 | 0.0068 |
| Model | E | H |
|---|---|---|
| ML-HMT | 0.0218 | 0.1267 |
| ML+P-HMT | 0.0037 | 0.0876 |
| VB-HMT | 0.0062 | 0.0963 |
| VBS-HMT | 0.0012 | 0.0068 |
Parameter estimation performance of the models applied to the tree structured data
| Model | E | H |
|---|---|---|
| ML-HMT | 0.0218 | 0.1267 |
| ML+P-HMT | 0.0037 | 0.0876 |
| VB-HMT | 0.0062 | 0.0963 |
| VBS-HMT | 0.0012 | 0.0068 |
| Model | E | H |
|---|---|---|
| ML-HMT | 0.0218 | 0.1267 |
| ML+P-HMT | 0.0037 | 0.0876 |
| VB-HMT | 0.0062 | 0.0963 |
| VBS-HMT | 0.0012 | 0.0068 |
At the testing step, we made use of the second set of 30 artificial lineage trees which were obtained using the same set of parameters θ used for generating the training set.
For the performance comparisons of the four models, at the testing step, we need to determine the percentage of correct predicted states of nodes.
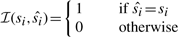
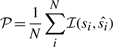
The trained models were applied to the testing set and the 𝒫 calculated for all four models. In Figure 3, we compare the performances of the four algorithms, varying the size of the training data. The aim here is to predict the unobserved cell type at all levels within each lineage tree in the test set as accurately as possible. From this figure it can be seen that, for the largest training set the models predicted the types of cell at all levels with high accuracy. The ML-HMT had the poorest performance. VB-HMT and the ML-HMT with supplementary prior information had similar behaviour and are superior to the simple ML approach. VBS-HMT had the best performance compared with the previous two models mentioned above. The VBEMS estimates obtained from the set of 30 stem cell lineage trees were very good as the produced posterior distributions are tight distributions (see Supplementary Material) and because the estimation of the full posterior captures most of the model uncertainties as opposed to a point estimate of the model-parameter posterior in the ML case. The use of synthetic data permitted a true test of the models since the information on the types of cells at all cell division levels within each lineage tree are known. For real lineage trees, such cell-type information at intermediate levels are generally unknown. It is to be noted that the resulting VBEMS algorithm generalizes the standard EM algorithm and its convergence is guaranteed (Attias, 2000; Beal and Ghahramani, 2003). The computational cost for the VB with smoothed probabilities approach is of the same order to that of the ML approach (Dasgupta and Carin, 2006).
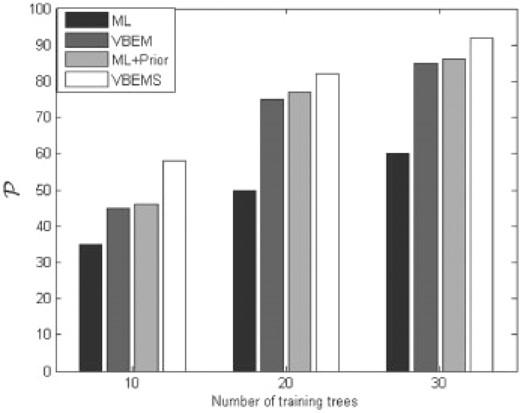
Model prediction performance with varying amounts of training data. Three blocks of bar diagrams have been produced showing the performance of the ML-HMT, VB-HMT, ML+P-HMT and VBS-HMT for a different size of training dataset: 10, 20, 30 lineage trees. The vertical axes shows the percentage of predicted cells type that have been right.
After model validation and comparison to other models in the literature using synthetic data, we applied the VBS-HMT model to the real stem cell lineage trees. The outcome of the time-lapse experiment consists of a dataset of 30 stem cell lineage trees in which the expression of SSEA3 can only be observed at the leaf and the root levels, i.e. x1 and xleaf are known. We estimated the model parameters θ = [π, vec(P), vec(C)]T using the VBEMS algorithm applied to the set of incomplete lineage trees. Subsequently, we predicted the presence or absence of SSEA3 expression at the unobserved positions within the trees, see Figure 4. In the biological context, π represents the probabilities of the root cell to be in one of the three possible states, i.e. SSEA3Positive, SSEA3Negative and dead, Pij expresses the probabilities of transitions from one of the possible states to another and Cjh expresses the probabilities of a cell of a particular state to be true stem cell, differentiated cell or dead cell. In several lineage trees, our model predicted that SSEA3Negative cells give rise to SSEA3Positive progeny. This suggests that NTERA2 stem cells could regain SSEA3 expression, i.e. the transition from SSEA3Negative to SSEA3Positiveis possible. This transition event has been validated by the real stem cell experiment in which a percentage of the root cells that were SSEA3Negative stem cells produced SSEA3Positive progeny.
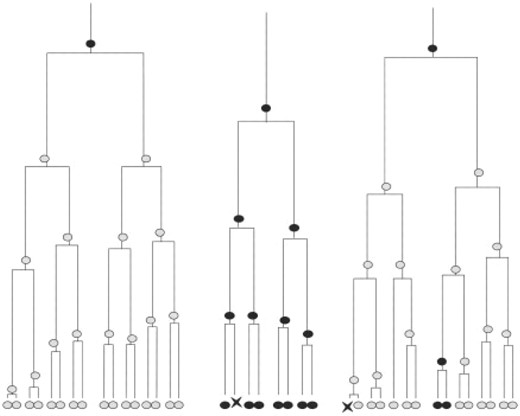
Diagram representing experimental stem cell lineage trees where light grey cells at the leaf level are observed positive definite, black cells at the root and the leaf levels are observed negative definite, the X shape cells are observed dead cells, the light grey cells at the intermediate levels are predicted SSEA3Positive cells, and the black cells at the intermediate levels are predicted SSEA3Negative cells.
Our proposed model, VBS-HMT, enables interpretation of data from real-time single cell analysis. Combining time-lapse analysis of single cells and the VBS-HMT model, it is possible to predict the unvisualized event of SSEA3 negative cells converting to the SSEA3 positive state. The VBS-HMT model predicts that the conversion of SSEA3 negative cells to the SSEA3 positive state occurs prior to the first cell division in 35% of cases. Culture conditions and numerous compounds affect the stability of cell populations, either inducing cell differentiation or stabilizing the cell population in its current state. Determining the kinetics and probability with which cells fluctuate between discrete states is important for understanding the physical properties of cells. Resolving this with the use of single cell analysis helps to identify if a cell population is functionally homogeneous or consists of different cell types. This information can in turn be used to more accurately direct the differentiation of stem cells to desired cell types. Utilization of the VBS-HMT model is not confined to the study of stem cell populations but can prove useful in the study of lineage selection during cell differentiation. Given data from a time-lapse analysis, the VBS-HMT model can help to predict when differentiating sister cells and progeny diverge with respect to cell type (i.e. mesoderm versus ectoderm).
Overall, the model enables us to predict decision events which are difficult to visualize. The results of the model simulations can then be used to generate testable hypothesis.
7 CONCLUSIONS
In this article, we developed the VBS-HMT model and applied it to incomplete tree structured data. The model proved to be superior to other models in the literature when tested on the prediction of the type of cells at each division level within a lineage tree as well as on the estimation of model parameters. We succeeded in confronting the underflow problems by combining the variational Bayesian method with the upward–downward algorithm with smoothed probabilities as an E-step in the EM context. The resulting algorithm was demonstrated to have superior performance over the competing approaches and was applied to the real stem cells lineage modelling problem. The VBS-HMT model provides the means to objectively predict a cell's phenotype from knowing the phenotype of the cells at the root and leaf level within the cell lineage tree. It is important to note that the proposed inference algorithm is able to predict novel behaviours based on incomplete data, which are not directly observable. These predictions can subsequently be validated by targeted experiments
Funding: Engineering and Physical Sciences Research Council (EPSRC).
Conflict of Interest: none declared.
REFERENCES
Author notes
Associate Editor: Olga Troyanskaya


{kind=link}
{kind=link}
{kind=link}
{kind=link}