




Abstract
Recent studies have shed light on the potential of circular RNA (circRNA) as a biomarker for disease diagnosis and as a nucleic acid vaccine. The exploration of these functionalities requires correct circRNA full-length sequences; however, existing assembly tools can only correctly assemble some circRNAs, and their performance can be further improved. Here, we introduce a novel feature known as the junction contig (JC), which is an extension of the back-splice junction (BSJ). Leveraging the strengths of both BSJ and JC, we present a novel method called JCcirc (https://github.com/cbbzhang/JCcirc). It enables efficient reconstruction of all types of circRNA full-length sequences and their alternative isoforms using splice graphs and fragment coverage. Our findings demonstrate the superiority of JCcirc over existing methods on human simulation datasets, and its average F1 score surpasses CircAST by 0.40 and outperforms both CIRI-full and circRNAfull by 0.13. For circRNAs below 400 bp, 400–800 bp, 800 bp–1200 bp and above 1200 bp, the correct assembly rates are 0.13, 0.09, 0.04 and 0.03 higher, respectively, than those achieved by existing methods. Moreover, JCcirc also outperforms existing assembly tools on other five model species datasets and real sequencing datasets. These results show that JCcirc is a robust tool for accurately assembling circRNA full-length sequences, laying the foundation for the functional analysis of circRNAs.
INTRODUCTION
Research on circular RNAs (circRNAs) has become a very important topic in recent years, including the aspects of prediction, sequence assembly and their association with diseases [1, 2]. CircRNA forms covalently closed loops through back-splicing [3] and exists in diverse organisms [4–6]. Moreover, circRNA is associated with numerous human diseases, including inflammatory skin diseases [7], hepatocellular carcinoma [8], Alzheimer’s disease [9] and others [10]. One prominent mechanism through which circRNA functions is by acting as a microRNA sponge, thereby regulating the expression of target genes [11]. Furthermore, circRNA exhibits protein interactions and can also translate proteins [12, 13].
The high sequence similarity between circRNA and mRNA presents a substantial challenge to accurately reconstruct the full-length sequences of circRNA using prediction methods. Meanwhile, circRNA exhibits multiple isoforms arising from alternative splicing [14]. To achieve accurate full-length sequences of circRNA, several algorithms have been developed, including CircAST [15], circseq_cup [16], CIRI-full [17] and circRNAfull [18], which utilize short reads sequencing data for sequence assembly. CircAST utilizes exon annotation information as a reference and constructs multiple splice graphs as circRNA models to reconstruct full-length sequences. However, its dependence on annotation information during the assembly process leads to the exclusion of intronic or intergenic circRNAs. In contrast, circseq_cup is a distinctive method that integrates circRNA prediction and assembly results without requiring additional circRNA prediction information as input. It relies on fusion junction sites and their corresponding paired-end RNA-seq reads for sequence assembly. circseq_cup does not have advantages regarding the sensitivity of circRNA identification compared to other current available methods, and as a result, it does not have advantages regarding the sensitivity of circRNA assembly. CIRI-full introduces a novel feature known as reverse overlap (RO), and by integrating both back-splice junction (BSJ) and RO features, CIRI-full reconstructs full-length circRNAs and improves the identification of low-abundance circRNAs. However, it is more suitable for longer sequencing reads (>250 or 300 bp). circRNAfull utilizes chimeric alignment from STAR aligners to reconstruct circRNA sequences. It focuses on identifying exon-skipping events rather than merely connecting all exons within the splicing site, resulting in an enhanced assembly rate. However, its performance is influenced by the read length and the insertion length, and it cannot reconstruct circRNAs located in intergenic regions.
Through the evaluation of existing assembly methods, we have summarized several challenges they encounter: (i) low assembly correct rates; (ii) limited capability to assemble long circRNA sequences; (iii) some methods exclusively focus on exonic circRNA. To address these challenges, this paper presents JCcirc, a tool specifically designed to assemble high-quality full-length circRNA sequences. Compared to existing methods, JCcirc introduces a novel feature called JC to enhance assembly accuracy and generate longer assembled sequences. JCcirc aligns JC and BSJ to the circRNA genome sequence and builds splice graphs based on shared JC and BSJ between aligned fragments, then obtains candidate full-length circRNA sequences after filtering low-confidence fragments based on fragment coverage. In this study, diverse evaluation metrics were employed to assess the performance of the assembly methods. As a result, JCcirc demonstrates an average precision of 0.70, an average recall of 0.50 and an average F1 score of 0.58 on human simulation data; its F1 score surpasses CircAST by 0.40 and outperforms both CIRI-full and circRNAfull by 0.13. Furthermore, for sequences within the length ranges of below 400 bp, 400–800 bp, 800–1200 bp and above 1200 bp, JCcirc achieves average correct assembly rates of 0.68, 0.46, 0.3 and 0.12, respectively. Moreover, JCcirc also surpasses existing assembly tools in other model organisms and real sequencing datasets.
RESULTS
It is still a challenge to assemble circRNA full-length sequences from short reads
We conducted a comprehensive evaluation of three assembly methods, namely CircAST, CIRI-full and circRNAfull, using simulated datasets of human circRNA. We did not use ‘circseq_cup’ because it relies on the quality information of sequencing reads, which was not provided in the simulated data. Among the three methods we used, CircAST exhibited the highest precision (average: 0.87) but the lowest recall (average: 0.1), resulting in a comparatively lower F1 score (average: 0.18) compared to the other two methods (Table 1). This can be attributed to the stringent threshold for BSJ employed by CircAST, which is set at the default value of 10. CIRI-full and circRNAfull demonstrated similar performance on 10 datasets (average F1 score, CIRI-full: 0.45, circRNAfull: 0.46).
Results of existing assembly methods on human simulated circRNA datasets
| Data | Precision (%) | Recall (%) | F1 score (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| M1 | M2 | M3 | M1 | M2 | M3 | M1 | M2 | M3 | |
| D10150 | 96.35 | 57.60 | 61.73 | 0.39 | 28.51 | 29.25 | 0.78 | 38.14 | 39.69 |
| D20150 | 92.79 | 62.45 | 64.28 | 3.83 | 33.91 | 35.01 | 7.35 | 43.96 | 45.33 |
| D30150 | 90.30 | 64.13 | 63.79 | 7.13 | 36.03 | 36.58 | 13.22 | 46.14 | 46.50 |
| D50150 | 87.16 | 66.45 | 62.70 | 11.16 | 38.74 | 37.32 | 19.79 | 48.94 | 46.79 |
| D70150 | 87.04 | 66.94 | 61.82 | 14.33 | 39.89 | 37.22 | 24.61 | 49.99 | 46.47 |
| D100150 | 84.74 | 67.36 | 60.50 | 16.67 | 40.73 | 36.95 | 27.86 | 50.76 | 45.88 |
| D30075 | 77.28 | 55.53 | 61.67 | 20.50 | 27.42 | 36.33 | 32.40 | 36.71 | 45.72 |
| D30100 | 81.18 | 59.09 | 62.89 | 16.43 | 31.40 | 36.98 | 27.33 | 41.00 | 46.57 |
| D30125 | 86.46 | 61.28 | 63.55 | 10.96 | 33.68 | 36.81 | 19.45 | 43.47 | 46.62 |
| D30150 | 90.30 | 64.13 | 63.79 | 7.13 | 36.03 | 36.58 | 13.22 | 46.14 | 46.50 |
| D30200 | 87.64 | 65.68 | 65.29 | 2.41 | 37.59 | 34.19 | 4.69 | 47.81 | 44.88 |
| Data | Precision (%) | Recall (%) | F1 score (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| M1 | M2 | M3 | M1 | M2 | M3 | M1 | M2 | M3 | |
| D10150 | 96.35 | 57.60 | 61.73 | 0.39 | 28.51 | 29.25 | 0.78 | 38.14 | 39.69 |
| D20150 | 92.79 | 62.45 | 64.28 | 3.83 | 33.91 | 35.01 | 7.35 | 43.96 | 45.33 |
| D30150 | 90.30 | 64.13 | 63.79 | 7.13 | 36.03 | 36.58 | 13.22 | 46.14 | 46.50 |
| D50150 | 87.16 | 66.45 | 62.70 | 11.16 | 38.74 | 37.32 | 19.79 | 48.94 | 46.79 |
| D70150 | 87.04 | 66.94 | 61.82 | 14.33 | 39.89 | 37.22 | 24.61 | 49.99 | 46.47 |
| D100150 | 84.74 | 67.36 | 60.50 | 16.67 | 40.73 | 36.95 | 27.86 | 50.76 | 45.88 |
| D30075 | 77.28 | 55.53 | 61.67 | 20.50 | 27.42 | 36.33 | 32.40 | 36.71 | 45.72 |
| D30100 | 81.18 | 59.09 | 62.89 | 16.43 | 31.40 | 36.98 | 27.33 | 41.00 | 46.57 |
| D30125 | 86.46 | 61.28 | 63.55 | 10.96 | 33.68 | 36.81 | 19.45 | 43.47 | 46.62 |
| D30150 | 90.30 | 64.13 | 63.79 | 7.13 | 36.03 | 36.58 | 13.22 | 46.14 | 46.50 |
| D30200 | 87.64 | 65.68 | 65.29 | 2.41 | 37.59 | 34.19 | 4.69 | 47.81 | 44.88 |
M1: CircAST, M2: CIRI-full, M3: CircRNAfull.
Results of existing assembly methods on human simulated circRNA datasets
| Data | Precision (%) | Recall (%) | F1 score (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| M1 | M2 | M3 | M1 | M2 | M3 | M1 | M2 | M3 | |
| D10150 | 96.35 | 57.60 | 61.73 | 0.39 | 28.51 | 29.25 | 0.78 | 38.14 | 39.69 |
| D20150 | 92.79 | 62.45 | 64.28 | 3.83 | 33.91 | 35.01 | 7.35 | 43.96 | 45.33 |
| D30150 | 90.30 | 64.13 | 63.79 | 7.13 | 36.03 | 36.58 | 13.22 | 46.14 | 46.50 |
| D50150 | 87.16 | 66.45 | 62.70 | 11.16 | 38.74 | 37.32 | 19.79 | 48.94 | 46.79 |
| D70150 | 87.04 | 66.94 | 61.82 | 14.33 | 39.89 | 37.22 | 24.61 | 49.99 | 46.47 |
| D100150 | 84.74 | 67.36 | 60.50 | 16.67 | 40.73 | 36.95 | 27.86 | 50.76 | 45.88 |
| D30075 | 77.28 | 55.53 | 61.67 | 20.50 | 27.42 | 36.33 | 32.40 | 36.71 | 45.72 |
| D30100 | 81.18 | 59.09 | 62.89 | 16.43 | 31.40 | 36.98 | 27.33 | 41.00 | 46.57 |
| D30125 | 86.46 | 61.28 | 63.55 | 10.96 | 33.68 | 36.81 | 19.45 | 43.47 | 46.62 |
| D30150 | 90.30 | 64.13 | 63.79 | 7.13 | 36.03 | 36.58 | 13.22 | 46.14 | 46.50 |
| D30200 | 87.64 | 65.68 | 65.29 | 2.41 | 37.59 | 34.19 | 4.69 | 47.81 | 44.88 |
| Data | Precision (%) | Recall (%) | F1 score (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| M1 | M2 | M3 | M1 | M2 | M3 | M1 | M2 | M3 | |
| D10150 | 96.35 | 57.60 | 61.73 | 0.39 | 28.51 | 29.25 | 0.78 | 38.14 | 39.69 |
| D20150 | 92.79 | 62.45 | 64.28 | 3.83 | 33.91 | 35.01 | 7.35 | 43.96 | 45.33 |
| D30150 | 90.30 | 64.13 | 63.79 | 7.13 | 36.03 | 36.58 | 13.22 | 46.14 | 46.50 |
| D50150 | 87.16 | 66.45 | 62.70 | 11.16 | 38.74 | 37.32 | 19.79 | 48.94 | 46.79 |
| D70150 | 87.04 | 66.94 | 61.82 | 14.33 | 39.89 | 37.22 | 24.61 | 49.99 | 46.47 |
| D100150 | 84.74 | 67.36 | 60.50 | 16.67 | 40.73 | 36.95 | 27.86 | 50.76 | 45.88 |
| D30075 | 77.28 | 55.53 | 61.67 | 20.50 | 27.42 | 36.33 | 32.40 | 36.71 | 45.72 |
| D30100 | 81.18 | 59.09 | 62.89 | 16.43 | 31.40 | 36.98 | 27.33 | 41.00 | 46.57 |
| D30125 | 86.46 | 61.28 | 63.55 | 10.96 | 33.68 | 36.81 | 19.45 | 43.47 | 46.62 |
| D30150 | 90.30 | 64.13 | 63.79 | 7.13 | 36.03 | 36.58 | 13.22 | 46.14 | 46.50 |
| D30200 | 87.64 | 65.68 | 65.29 | 2.41 | 37.59 | 34.19 | 4.69 | 47.81 | 44.88 |
M1: CircAST, M2: CIRI-full, M3: CircRNAfull.
As anticipated, the performance of the assembly methods exhibited improvement with increasing read coverage. With a 10-fold increase in coverage, CircAST showed an increase in the F1 score by 0.27, while circRNAfull’s F1 score only increased by 0.06. In addition, we also observed a significant impact of read length on the performance of circRNA assemblers. Consistent with prior findings, CircAST demonstrated superior performance with shorter read lengths, achieving a higher F1 score at 75 bp (F1 score: 0.32) compared to 200 bp (F1 score: 0.04) [19]. Conversely, CIRI-full exhibited better performance in longer read lengths. Read length increased from 75 to 200 bp, resulting in a 0.11 increase in the F1 score for CIRI-full. circRNAfull shows less dependence on read length in terms of its performance.
We also conducted an analysis on human circRNA simulated datasets to determine the assembly number and assembly rate of circRNA sequences across different length ranges. As illustrated in Figure 1, all methods exhibited better performance on shorter sequences compared to longer ones. When comparing results for sequences below 400 bp to those above 1200 bp, the average correct assembly rates for CircAST, CIRI-full and circRNAfull decreased by 0.12, 0.55 and 0.33, respectively. For sequences shorter than 400 bp, CIRI-full demonstrated the highest correct assembly rate (average: 0.55) (Figure 1A), while for sequences longer than 400 bp, circRNAfull outperformed the other two methods (Figure 1B–D). Overall, circRNAfull exhibited a more stable performance across all sequence lengths. In addition, we also categorized the assembled circRNA sequences. The results, presented in Table 2, indicate that current methods perform more effectively in assembling exonic circRNAs but perform worse in exon–intron, intronic and intergenic circRNAs.
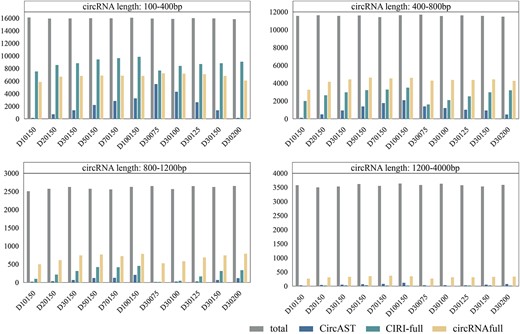
Assembly results of full-length circRNA sequences in different length intervals. The bar chart represents the number of correctly assembled circRNA sequences. The length intervals are as follows: (A) circRNA length between 100 bp and 400 bp, (B) circRNA length between 400 bp and 800 bp, (C) circRNA length between 800 bp and 1200 bp and (D) circRNA length between 1200 bp and 4000 bp.
Average proportions of circRNA assembly sequence categories on human simulated circRNA datasets
| Assembly method | Exonic | Exonic–intronic | Intronic | Intergenic |
|---|---|---|---|---|
| CircAST | 12.64% | 0.00% | 0.00% | 0.00% |
| CIRI-full | 42.20% | 0.92% | 0.72% | 1.16% |
| circRNAfull | 43.41% | 0.00% | 0.00% | 0.00% |
| Assembly method | Exonic | Exonic–intronic | Intronic | Intergenic |
|---|---|---|---|---|
| CircAST | 12.64% | 0.00% | 0.00% | 0.00% |
| CIRI-full | 42.20% | 0.92% | 0.72% | 1.16% |
| circRNAfull | 43.41% | 0.00% | 0.00% | 0.00% |
Average proportions of circRNA assembly sequence categories on human simulated circRNA datasets
| Assembly method | Exonic | Exonic–intronic | Intronic | Intergenic |
|---|---|---|---|---|
| CircAST | 12.64% | 0.00% | 0.00% | 0.00% |
| CIRI-full | 42.20% | 0.92% | 0.72% | 1.16% |
| circRNAfull | 43.41% | 0.00% | 0.00% | 0.00% |
| Assembly method | Exonic | Exonic–intronic | Intronic | Intergenic |
|---|---|---|---|---|
| CircAST | 12.64% | 0.00% | 0.00% | 0.00% |
| CIRI-full | 42.20% | 0.92% | 0.72% | 1.16% |
| circRNAfull | 43.41% | 0.00% | 0.00% | 0.00% |
In summary, our findings suggest that different methods are suitable for different datasets. However, the overall performance of existing methods is hindered by imbalanced precision and recall, or low precision and recall. Except for CIRI-full on D100150, the F1 scores for all other datasets remain below 0.5. Furthermore, although all methods achieve relatively high assembly rates for short circRNA sequences, the highest correct assembly rate is only 0.55. In addition, correctly assembling non-exonic circRNA sequences continues to pose a significant challenge.
The impact of junction contigs from different de novo assemblers
In this study, we present a novel method called JCcirc, which incorporates JC and enables the efficient reconstruction of all types of circRNA full-length sequences and their alternative isoforms using splice graphs and fragment coverage.
JC is assembled by a de novo transcript assembler. To analyze the effect of JC assembled by different de novo assemblers on JCcirc, we compared the performance of four assemblers (Trinity [20], SOAPdenovo [21], SPAdes [22] and ABySS [23]) combined with JCcirc on simulated circRNA datasets. These datasets include four animal species (human, mouse, fish and fly) and two plant species (Arabidopsis thaliana and rice) (see details in Method). The selection of these assemblers was based on their superior performance in previous evaluations conducted by Holzer et al. [24].
Our results showed that Trinity and SPAdes exhibited similar average F1 scores in each species and slightly outperformed SOAP and ABySS. The average F1 score of ABySS is the lowest in each species (as depicted in Figure 2). For example, on the mouse datasets, the average F1 scores for Trinity, SPAdes, SOAP and ABySS were 0.57, 0.55, 0.57 and 0.52, respectively. For human datasets, the precision and recall of SOAP, SPAdes and Trinity performed variably on different datasets. However, for datasets of other species, the precision and recall rankings of the four assemblers combined with JCcirc, from high to low, were Trinity, SPAdes, SOAP and ABySS. This trend was consistent with the changes in F1 scores (Figure S1).
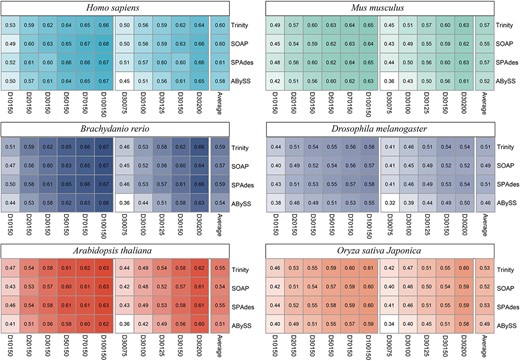
Performance comparison of four de novo transcript assemblers combined with JCcirc. The numbers in each grid cell indicate the F1 score of the corresponding assembly method. The last column ‘Average’ of each subgraph represents the average of F1 score for 10 datasets on each species.
It is evident that the choice of de novo assemblers only have a slight impact on the final assembly results in JCcirc, except for ABySS. This can be attributed to the role played by JC in the circRNA sequence, which could support fragments that are not covered by BSJ or add the coverage of these fragments (Figure S2). The contribution of JC to sequence assembly is primarily through aligned fragments rather than the entire JC sequence. Hence, the JC sequence obtained from different de novo assemblers has a slight influence on the final results.
In addition, the data size of assembly results obtained by the de novo assemblers can affect the runtime of JCcirc. Taking the circRNA simulation data D50150 of mouse as an example, Trinity has the largest data size of assembly results and runtime is 647 s, while ABySS has the smallest data size and runtime is 613 s (Table S1). This can also be an index for selecting an assembly tool. Here, we recommend utilizing Trinity for de novo assembly in JCcirc, and all subsequent analyses were performed using Trinity.
The effect of junction contigs on JCcirc
Our findings demonstrate that the inclusion of JC significantly improves the performance of JCcirc on human simulated datasets. F1 scores increased significantly when JC was added, with the maximum improvement of 0.27 observed in data D30075 (Figure 3A). Moreover, the precision and recall of the assembly results for all 10 datasets exhibited the same trend (Figure S3). However, it is worth noting that the positive effect of JC is influenced by the read coverage and length. As the read coverage and length increase, the impact of JC on the assembly results gradually diminishes (Figure 3A). In addition, the incorporation of JC into the assembly process benefits the assembly of longer circRNA sequences. Across the 10 datasets, we observed the median length of the 10 datasets increased by 4 to 18 bp in the assembled sequences (Figure 3B, Table S2). Notably, the highest increase of 18 bp was observed when the read coverage was the lowest (D10150, with a coverage of 10).
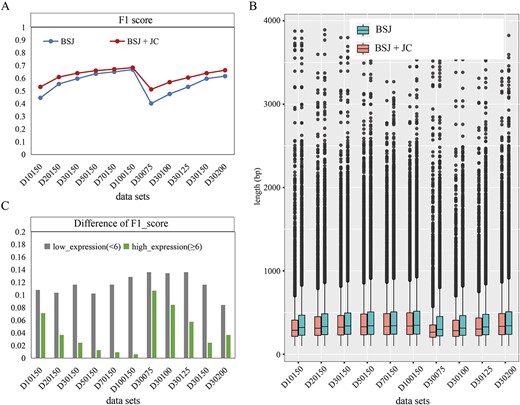
The impact of JC on the assembly performance of JCcirc across 10 simulated human circRNA datasets. (A) F1 scores of the assembly results obtained from JCcirc with and without JC. (B) The length distribution of circRNA assembly sequences obtained from JCcirc with and without JC. (C) The effects of incorporating JC on the assembly performance of low-expression and high-expression circRNA. The bar graph represents the difference in F1 scores between the inclusion and exclusion of JC.
Moreover, we compared the effects of incorporating JC on assembling low-expression circRNA (number of BSJ < 6) and high-expression circRNA (number of BSJ ≥ 6). The results indicate that JC has a more pronounced effect on improving the assembly rate of low-expression circRNA compared to high-expression circRNA. On human datasets, the average F1 score of low-expression circRNA increased by 0.12, whereas the average F1 score of high-expression circRNA only increased by 0.04 (Figure 3C, Figure S3C and D). For low-expression circRNA, there are few BSJs available for sequence assembly, and the proportion of JC is similar to BSJ, which means that JC is not easily filtered out in subsequent steps. By incorporating JC, we can effectively increase the total information available for assembly, leading to a noticeable improvement in correct assembly rates. However, high-expression circRNAs have a smaller proportion of JC compared to BSJ, making them more susceptible to being filtered out in subsequent steps. Therefore, for high-expression circRNAs, the impact of adding JC is relatively minor compared to the significant benefits for low-expression circRNAs.
The impact of linear RNA reads on assembly results
Current research on circRNA often constructs circRNA libraries using RNase R to eliminate linear RNA and enrich circRNA. However, certain linear RNAs exhibit resistance to RNase R [25], and circRNA enrichment libraries frequently contain a mixture of circRNA reads and linear RNA reads. To assess the impact of linear RNA reads on assembly tools, we compared their performance on simulated human circRNA datasets with and without linear RNA reads.
As depicted in Figure S4, linear RNA reads have a minor influence on JCcirc, with differences in F1 score, precision and recall not exceeding 0.02 across all datasets. Thus, the residual linear RNA reads in the circRNA library have negligible consequences on the assembly results of JCcirc. We also examined the impact of linear RNA reads on other existing circRNA full-length assemblers (Figure S5). Interestingly, linear RNA reads had a minor influence on CIRI-full and circRNAfull but exerted a significant impact on CircAST. The impact of linear RNA reads on the circRNA assembly methods suggests that the results of real sequencing data may include false-positive sequences.
JCcirc outperforms on human simulated circRNA datasets
We next compared the performance of JCcirc with existing circRNA sequence assemblers on human circRNA simulated datasets. JCcirc exhibited the highest average F1 score of 0.58, primarily due to its superior recall. The average F1 score of JCcirc was higher by 0.13 compared to CIRI-full and circRNAfull, and 0.40 compared to CircAST (Figure 4A, Table S3). However, CircAST achieved the highest average precision at 0.87, surpassing JCcirc by 0.17. This advantage can be attributed to the stringent threshold for BSJ employed by CircAST, while JCcirc does not set a threshold for BSJ.

Performance of four assembly methods for assembling circRNA full-length sequences on simulated human datasets. (A) Precision and recall values of the assembly methods. (B) Overlap of assembly results from different circRNA full-length sequence assemblers. (C) The assembly rate of circRNA full-length sequences by four assembly methods in different length intervals. (D) Assembly results of alternative splicing circRNA. The bar chart represents the number of correctly assembled circRNA full-length sequences for each method, while the line chart depicts the assembly rate of alternative splicing circRNA.
In addition, JCcirc consistently demonstrated a superior correct assembly rate in every length interval compared to existing methods (Figure 4C). Previous analysis revealed that CIRI-full performed best for sequences shorter than 400 bp, while circRNAfull was optimal for sequences longer than 400 bp. Here, JCcirc achieved an average correct assembly rate of over 0.68 for circRNA sequences shorter than 400 bp, increased by 0.13 compared to CIRI-full. Furthermore, compared to circRNAfull, the average correct assembly rates of JCcirc increased by 0.09, 0.04 and 0.03 in the length intervals of 400–800 bp, 800–1200 bp and above 1200 bp, respectively.
We also compared the circRNA sequences assembled by JCcirc and other methods. In total, CircAST, CIRI-full, circRNAfull and JCcirc correctly assembled 25 954, 66 492, 65 959 and 102 922 circRNA sequences, respectively (Figure 4B). Notably, the JCcirc’s results covered a significant proportion of the results assembled by other methods, accounting for 95.40% of CircAST, 93.00% of CIRI-full and 74.93% of circRNAfull. This indicates that the assembly results of JCcirc are more comprehensive.
Alternative splicing of circRNA was analyzed from each method, considering four types of alternative splicing: skipping exons (SE), retained intron (RI), alternative 5′ splice site (A5SS) and alternative 3′ splice site (A3SS). The presence of RI was negligible in all four assembly methods, while A5SS and A3SS exhibited a high assembly rate of detection by each method. JCcirc displayed the highest average assembly rate for both A5SS (62.97%) and A3SS (65.16%), while CIRI-full achieved the highest rate for SE (5.66%) (Figure 4D).
Previous results showed that the majority of correctly assembled circRNAs belong to exonic circRNA. On human simulated circRNA datasets, the average assembly rates of JCcirc for different circRNA types exceeded existing methods, exonic circRNA is 60.32%, exon–intron circRNA is 17.13%, intronic circRNA is 9.12% and intergenic circRNA is 8.56%.
Comparison of assembly results between JCcirc and other assembly methods in multiple species
In addition to its exceptional performance on human simulated datasets, JCcirc also has superiority over existing methods on five other species, as depicted in Figure S6. Across all analyzed species, JCcirc consistently achieves the highest recall and F1 scores, with precision second only to CircAST. Moreover, in the case of mouse, fish and fly, the average recall and F1 scores of circRNAfull were slightly superior to those of CIRI-full, while the average recall and F1 scores of CIRI-full were slightly higher than circRNAfull in rice and A. thaliana. circRNAfull exhibits superior performance in animal datasets, but it lags behind CIRI-full in plant datasets.
Benchmark real datasets
The standard for evaluating assembled circRNA sequences derived from real short-read RNA-seq data is utilizing circRNA-enriched long-read sequencing libraries obtained from the same cell or tissue. Long-read sequencing allows for the direct detection of RNA sequences longer than 1000 bp. In order to evaluate the assembly tools for circRNA full-length sequences, we utilized publicly available Illumina circRNA-seq data samples along with matching long-read circRNA-seq data (Nanopore) to ensure consistency (Table S5). These validation datasets included samples from mouse brain tissue and various human cell lines.
We assessed the performance of five circRNA sequence assemblers, JCcirc, CIRI-full, CircAST, circRNAfull and circseq_cup, on the real RNA-seq datasets. The performance of the assembly methods on the real datasets is consistent with the findings on the simulated datasets (Table S4). CircAST exhibited the highest precision (average: 0.81, Figure 5A), and JCcirc demonstrated the highest recall (average: 0.41, Figure 5B) and the highest F1 scores (average: 0.45, Figure 5C).
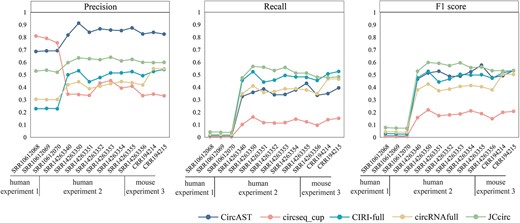
Performance of circRNA full-length sequence assembly methods on the real RNA-seq datasets. Results of the analysis on the 13 Illumina RNA-seq datasets. (A) precision, (B) recall and (C) F1 score.
Furthermore, the performance of CircAST, circseq_cup, CIRI-full and circRNAfull was notably influenced by the experimental datasets. circseq_cup demonstrated the highest precision in experiment 1 datasets (average: 0.79) while performing moderately in the experiment 2 datasets (average precision: 0.38) and experiment 3 datasets (average precision: 0.34) (Figure 5A). Excluding JCcirc, circRNAfull achieved the highest F1 scores in experiment 1 datasets (average: 0.04), CircAST obtained the highest F1 scores in experiment 2 datasets (average: 0.51) and CIRI-full obtained the highest F1 scores in experiment 3 datasets (average: 0.53). Besides, Figure 5 demonstrates that the recall and F1 scores of experiment 1 were significantly lower than the other two groups. This can be attributed to the low sequencing depth of the long-read sequencing data in experiment 1, which resulted in fewer reference circRNAs. In other words, some circRNAs may have been correctly assembled, but they could not be verified by reference circRNA sequences. Thus, experiment 1 exhibited lower recall and F1 values compared to the other two groups.
DISCUSSION
In recent years, researchers have increasingly recognized the significant role of circRNAs in various diseases [26], and the functional properties of circRNAs heavily rely on their sequence composition. To enhance the correct assembly rate of circRNA sequences, we have developed a novel assembly tool known as JCcirc. This tool integrates BSJ and JC to facilitate the correct assembly of circRNA full-length sequences. Our results demonstrated that JCcirc achieves a high assembly rate while maintaining high precision, surpassing other existing assembly methods. JCcirc also possesses the advantage of effectively assembling all types of circRNAs, including exonic, intronic, exonic–intronic and intergenic circRNAs.
JCcirc introduced non-BSJ for the first time during the assembly of full-length circRNA sequences, significantly enhancing the assembly performance. In this study, we assessed the potential for false positives resulting from including non-BSJ using circRNA simulated datasets that incorporated linear reads. It was observed that the negative impact of introduced non-BSJ on the assembly results was minimal, with the majority of these reads originating from circRNA sequences. This is because we searched for highly correlated reads with BSJ when introducing non-BSJ. We obtained JC through a de novo assembly tool, which served as a reliable bridge connecting BSJ and high-correlation non-BSJ. Furthermore, we aligned the JC to the circRNA genome sequences, disregarding fragments that failed to align. Therefore, while some non-BSJ were introduced, they rarely caused false positives.
In the analysis of real datasets, we used long-read sequencing data to validate the assembled circRNAs sequences. We used three sets of long-read sequencing data, employing two distinct experimental methods aimed at reducing error rates. The first approach is rolling circle amplification followed by nanopore long-read sequencing [27], and another approach utilizes rolling circular reverse transcription in conjunction with nanopore long-read sequencing [28, 29]. Both of these methods sequenced the reads after generating multiple replicates of transcripts, effectively lowering the sequencing error rate. Also, we employed two circRNA prediction methods: CIRI-long and isoCirc. CIRI-long generated cyclic consensus sequences (CCS) for each nanopore read, with CCS accuracy ranging from 92.6 to 98.1%. isoCirc detected tandem repeats within raw reads and generated consensus sequences, and the error rate drops to less than 5% when consensus sequences have more than four copies.
In the results of real datasets, JCcirc consistently maintained high recall and F1 scores across different datasets (Figure 6). However, the performance of other methods depends on the specific datasets. For instance, in experiment 3 datasets, which involved mouse brain tissue, the differences in F1 scores among the methods were smaller than in the experiment 2 datasets obtained from human cell lines. Interestingly, circseq_cup’s performance exhibited unexpected variations across different datasets. In experiment 1 datasets, its precision surpassed that of CircAST. Conversely, in experiment 2 and experiment 3 datasets, circseq_cup consistently exhibited the lowest precision and recall compared to other methods. Further investigation revealed that experiment 1 and experiment 2 datasets are both derived from human cell lines, with experiment 1 having shorter read lengths (100 bp) than experiment 2 (150 bp), but twice the coverage compared to experiment 2. It suggested that the sequencing depth significantly impacts the performance of circseq_cup.
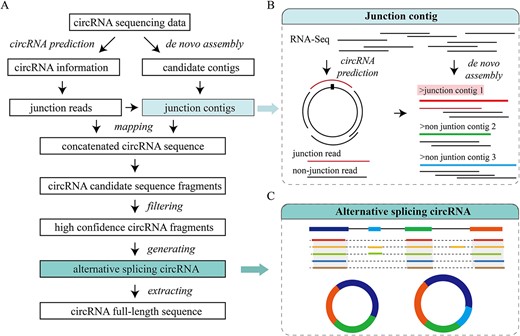
(A) Workflow of JCcirc. (B) Illustration of the junction contig. (C) Formation of alternative splicing circRNA.
Although JCcirc demonstrates superior performance in assembling circRNA sequences, the assembly of some specific circRNA types remains challenging, with the correct assembly rate for intronic and intergenic circRNAs still falling below 10%. Moreover, despite the utilization of JC in JCcirc to extend the length of assembled sequences, the correct assembly rate of sequences exceeding 1200 bp remains low. In this study, we introduced some non-BSJ through JC to assemble the full-length sequence of circRNA. Based on the JC, we can further expand some weakly correlated non-BSJ that are associated with highly correlated non-BSJ. These reads may prove beneficial for the assembly of long-sequence circRNAs. However, the probability of weakly correlated non-BSJ still derived from circRNA is lower than that of non-BSJ in JC, necessitating further filtering to ensure their reliability.
The coexistence of circRNA reads and linear RNA reads within sequencing data presents a significant challenge in the assembly of circRNA full-length sequences. Existing methods typically rely on BSJ for assembly, which restricts the length of the assembled sequences. In contrast, JCcirc introduces JC, which leverages non-BSJ to enhance the assembly rate of circRNA sequences. The inclusion of JC has indeed yielded improvements in the assembly rate, particularly for low-expression circRNAs. Nevertheless, there remains scope for further advancements in assembling long-sequence circRNAs and other circRNA types beyond exonic. It is still important to develop assembly tools for long-sequence circRNAs and non-exonic circRNAs.
METHOD
Datasets: simulated datasets
In order to address the limitation of CIRI_simulator, which focuses exclusively on simulating exonic circRNA sequencing reads, we have developed CircSimu based on CIRI_simulator [30]. CircSimu is capable of generating simulated sequencing data for four types of circRNA (exonic, exonic–intronic, intronic and intergenic). The source code for this software tool can be accessed at https://github.com/cbbzhang/JCcirc.
The utilization of simulated data allows for the acquisition of all information regarding circRNA, facilitating a comprehensive evaluation of assembly tools. To assess the performance of the assembly methods, we generated simulated data comprising paired-end reads of both circRNAs and linear RNAs. CircSimu was employed to generate the circRNA reads, and the linear RNA reads were generated using Polyester [31].
Two groups of circRNA reads were used to evaluate the performance of the assembly methods. The first group (G1) of simulated circRNA datasets comprises six datasets with varying sequencing depths (10×, 20×, 30×, 50×, 70×, 100×). All datasets in G1 have a read length of 150 bp and an average insert length of 450 bp with a standard deviation of 70 bp. The second group (G2) of simulated circRNA datasets consists of five datasets with different read lengths (75 bp, 100 bp, 125 bp, 150 bp and 200 bp). The sequence depth for G2 is set to 30×, while the average insert length and standard deviation of the insert length remain the same as in G1 (450 bp and 70 bp). The read lengths of the linear RNA simulated data in Polyester are the same as circRNA, with the ‘reads_per_transcript’ parameter set to 300 [32].
To assess the robustness of the assembly methods, we generated simulated circRNA datasets for six different model species using their reference genomes. The species included in the study were human (Homo sapiens, GRCh38/hg38), mouse (Mus musculus, GRCm38/mm10), fruit fly (Drosophila melanogaster, BDGP R6/dm6), zebrafish (Brachydanio rerio, GRCz11/danRer11), A. thaliana (Arabidopsis thaliana, TAIR10) and rice (Oryza sativa Japonica, IRGSP-1.0). Genome information for animals was obtained from UCSC, and genome information for plants was sourced from Ensembl.
Datasets: real datasets
Moreover, we collected three groups of human and mouse datasets from the Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra) and the National Genomics Data Center (https://ngdc.cncb.ac.cn/). Each group included both Illumina RNA-seq data (short reads) and Nanopore sequencing data (long reads) derived from the same experimental samples. The reference genomes for human (GRCh38/hg38) and mouse (GRCm38/mm10) were obtained from UCSC. The detailed information of these datasets can be found in Table S5.
Description of JCcirc
Existing assembly methods heavily rely on BSJ for the reconstruction of circRNA full-length sequences. However, short reads pose challenges in achieving high assembly accuracy and effectively assembling long sequences. To improve the correct assembly rate, we have developed JCcirc. This tool introduces the concept of JC, an extension of the BSJ, and integrates both BSJ and JC to reconstruct full-length circRNA sequences. JCcirc is accessible at https://github.com/cbbzhang/JCcirc and consists of four steps, which are visually depicted in Figure 6.
First step: extraction of BSJ and JC
BSJ was predicted by circRNA prediction algorithms, and JC was assembled by de novo assembly algorithms from RNA-seq data. The assembly contigs are classified into two distinct groups: JC, assembled from both BSJ and non-BSJ (Figure 6B); and non-junction contigs, assembled solely from non-BSJ.
Second step: alignment of BSJ and JC
Once the circRNA junction sites are identified, we extract the genomic sequence between the two sites. The two genomic sequences are then concatenated, forming a single sequence where the junction represents a back-splicing site. We employ BWA with default parameters to align the BSJ and JC to this concatenated reference sequence. The alignment process yields a SAM file containing the mapping results. Based on these mapping results, we obtain candidate fragments related to the full-length sequences of circRNAs.
Third step: filtering of low-confidence fragments
First of all, we performed a fine-tuning process for fragment locations based on the provided annotation file. When the distance between a fragment site and an exon site was ≤20 nt, we fine-tuned the fragment site to the exon site. For two fragments situated within the same exon, we generated a new fragment with the start site set as the smallest among the fragments’ sites and the end site set as the largest.
Subsequently, we applied several criteria to merge or delete fragments based on the following rules: (i) Fragment shorter than 20 nt was excluded. (ii) In cases where one fragment was entirely covered by another fragment, we retained the fragment with higher coverage. (iii) Fragments with partial overlap were merged to form a longer fragment. (iv) Fragments with a coverage of at least 3 or coverage equal to or greater than the average coverage minus 2 were retained. By implementing these filtering criteria, we ensured the selection of highly reliable fragments for subsequent reconstruction.
Fourth step: generation of alternative splicing circRNA
Primarily, we generate potential splice graphs based on shared BSJ IDs or JC IDs, where candidate fragments serve as nodes, and BSJ and JC represent edges in these graphs. If the same BSJ or JC supports two fragments, these two nodes may be adjacent. We then generate the longest full-length sequences according to the longest splice graph. Next, we filter out low-confidence fragments based on fragment coverage to identify reliable splice graphs as potential circRNA alternative splicing isoforms. Fragments with low coverage are excluded as they may not provide sufficient support for multiple circRNAs. Given that our sequence alignment primarily relies on BSJ and JC, higher coverage at both ends of the sequence compared to the middle region. Assuming a circRNA has N alignment fragments, we make the following assumptions:
(i) The coverage of fragment 1 is approximately equal to the coverage of fragment N, the coverage of fragment 2 is approximately equal to the coverage of fragment N − 1, and so forth.
(ii) The coverage of fragment 2 is less than that of fragment 1.
Based on these assumptions, we apply the following rules:
If the coverage of two corresponding fragments, i and j, differ by three or more (coveragei − coveragej ≥ 3), and (coveragej + D) < coveragej + 1 and (coveragej + D) < coveragej − 1, we exclude fragment j in circRNA isoforms. D can be set to 0, 1 or 2; a larger value means stricter screening. This parameter can also be adjusted based on the intron length of the reference genome; longer introns could set smaller values.
When N is an odd number, we compare the coverage of the median fragment with the coverage of the fragments median − 1 and median + 1. If coveragemedian < coveragemedian − 1/2 and coveragemedian < coveragemedian + 1/2, we exclude fragment median in circRNA isoforms.
Finally, if the length of the assembled sequence is greater than 5000 bp, we delete the sequence.
Prediction of circRNA and assembly of circRNA full-length sequences
To analyze the sequencing data obtained from experimental samples, we employed several existing tools. For RNA-Seq data (short reads), we utilized CIRI2 (version 2.0.6) [30] in combination with BWA (version 0.7.17) [33], CIRCexplorer2 (version 2.3.0) [34] in combination with BWA, and DCC (version 0.5.0) [35] in combination with STAR (version 2.7.6a) [36] to predict circRNAs. The circRNAs identified by all three methods simultaneously were considered for subsequent assembly. For Nanopore sequencing data, we employed isoCirc (version 1.0.2) [27] or CIRI-long (version 1.0.1) [29] to predict circRNA sequences. isoCirc is more suitable for long reads longer than 1000 bp, while CIRI-long is more suitable for long reads shorter than 1000 bp.
To generate the circRNA full-length sequences, we utilized five different assemblers: CircAST combined with Tophat2 (version 2.1.0) [37], circseq_cup (version 1.0) combined with Tophat2, CIRI-full (version 2.0) combined with BWA, circRNAfull combined with STAR, and our proposed JCcirc. All of these software tools were executed using their default parameters.
Evaluation metrics
In order to assess the correctness of the assembled circRNA sequences, we conducted comparisons with simulated sequences or sequences predicted from third-generation sequencing. We considered an assembly sequence to be correct if this sequence has the same number of fragments as the reference sequence, and each fragment of the circRNA assembly sequence aligned perfectly with the corresponding reference fragment. A tolerance of up to 6 bp displacement was allowed between the start and end sites of each fragment and the reference fragment sites.
To evaluate the performance of circRNA full-length sequence assemblers, we utilized several metrics, namely precision, recall and F1 score. Higher scores indicate superior performance across all of these metrics. The calculation formulas for these metrics are defined as follows:
where TP indicates true positives, FP is false positives, TN refers to true negatives and FN represents false negatives.
We have developed an efficient tool named JCcirc, which is used for reconstructing full-length circRNAs through integrated junction contigs.
JCcirc consistently outperforms other existing methods on simulated datasets of multiple model organisms and real datasets.
In comparison to other methods, JCcirc exhibits superior assembly rates for all types of circRNAs, including exonic circRNAs, intronic circRNAs, exonic–intronic circRNAs and intergenic circRNAs.
FUNDING
This work is partly supported by the Key Research and Development Project of Guangdong Province under grant no. 2021B0101310002, the National Science Foundation of China under grant no. 62272449, the Strategic Priority CAS Project under grant no. XDB38050100, the National Key Research and Development Program of China under grant no. 2021YFF1200104, the Shenzhen Basic Research Fund under grant nos. RCYX20200714114734194, KQTD20200820113106007 and ZDSYS20220422103800001, and by the Youth Innovation Promotion Association (Y2021101), CAS to Yanjie Wei.
DATA AVAILABILITY
All required links or identifiers for our data are present in the manuscript and supplementary data.
Jingjing Zhang is a PhD student at the University of Chinese Academy of Sciences, Beijing, China. Her research interest is bioinformatics and circular RNAs.
Huiling Zhang is currently a lecturer at South China Agricultural University. Her research interest lies in the fields of big data, machine learning, computational biology and bioinformatics.
Zhen Ju is a PhD student at the University of Chinese Academy of Sciences, Beijing, China. His current research interests include heterogeneous computing and high-performance computing.
Yin Peng is an associate professor in department of Pathology at Shenzhen University, China. Her long-standing research interests lie in elucidating the molecular mechanisms of cell signaling transduction pathway underlying tumorigenesis.
Yi Pan is a Chair Professor and the Dean of the College of Computer Science and Control Engineering, Shenzhen Institute of Advanced Technology, Chinese Academy of Sciences, China, and a Regents’ Professor Emeritus with Georgia State University, USA. His current research interests include bioinformatics and health informatics using big data analytics, cloud computing, and machine learning technologies.
Wenhui Xi is an associate professor at Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences. His research interests include bioinformatics and molecular simulation of proteins and peptides.
Yanjie Wei is a professor and the director in Center for High Performance Computing, Shenzhen Institute of Advanced Technology, Chinese Academy of Sciences. His research focuses on high performance computing and computational biology/bioinformatics.
References

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}