




Abstract
Protein remote homology detection is essential for structure prediction, function prediction, disease mechanism understanding, etc. The remote homology relationship depends on multiple protein properties, such as structural information and local sequence patterns. Previous studies have shown the challenges for predicting remote homology relationship by protein features at sequence level (e.g. position-specific score matrix). Protein motifs have been used in structure and function analysis due to their unique sequence patterns and implied structural information. Therefore, designing a usable architecture to fuse multiple protein properties based on motifs is urgently needed to improve protein remote homology detection performance. To make full use of the characteristics of motifs, we employed the language model called the protein cubic language model (PCLM). It combines multiple properties by constructing a motif-based neural network. Based on the PCLM, we proposed a predictor called PreHom-PCLM by extracting and fusing multiple motif features for protein remote homology detection. PreHom-PCLM outperforms the other state-of-the-art methods on the test set and independent test set. Experimental results further prove the effectiveness of multiple features fused by PreHom-PCLM for remote homology detection. Furthermore, the protein features derived from the PreHom-PCLM show strong discriminative power for proteins from different structural classes in the high-dimensional space. Availability and Implementation: http://bliulab.net/PreHom-PCLM.
INTRODUCTION
The protein remote homology detection is an essential step in many protein structure and function predictors [1, 2], revealing the evolutionary relationship among different organisms [3, 4]. Recent studies have demonstrated that it is possible to predict the 3D structures of proteins by using the evolutionary relationship inferred from the large-scale biological data by sequence analysis methods. For example, the HH-suite [5] has been employed by AlphaFold2 [6] and RoseTTAFold [7]. Therefore, more accurate remote homology detection methods are useful for studying protein structures and functions so as to understand disease mechanisms [7–9].
Remote homology detection is usually formalized as a classification or a retrieval problem (i.e. ranking problem). Several computational methods have been proposed, which can be divided into four groups: alignment methods, protein similarity network methods, ensemble learning methods and deep learning methods.
PSI-BLAST [10, 11] is a fundamental alignment method, which iteratively searches the homology sequences based on the position-specific score matrix (PSSM) [12]. Motived by this approach, methods based on different sequence profiles are proposed, such as HMMER [13, 14] and HHblits [5, 15]. They iteratively search the homology sequences based on Hidden Markov Model (HMM) profiles. MMseqs2 combines the short word (k-mer) match and the Smith–Waterman alignment algorithms for the large-scale metagenomic data analysis [16–18]. Its performance is comparable with PSI-BLAST requiring lower computational cost. However, traditional alignment methods that rely solely on sequence-level information struggle to accurately predict remote homology relationships, which can lead to nonhomologous proteins in their predictions [19, 20].
The protein similarity network provides structural information by constructing a structural-similarity-based weighted graph. The enrichment of Network Topological Similarity [21] is applied to sequence analysis and achieves better performance via building a structure similarity network. HITS-PR-HHblits [22] performs the PageRank and Hyperlink-Induced Topic Search on the similarity network to re-rank the results of PSI-BLAST. PL-search [23] constructs the double-link and profile-link networks, and further calculates the Jaccard score to filter the nonhomologous sequences caused by PSI-BLAST.
Ensemble learning is another way to improve predictive performance by learning multiple protein properties. ProtDec-LTR [24–26] integrates multiple ranking methods based on the Learning to Rank (LTR) [27] framework and obtains more accurate results. The LTR framework is a supervised framework for capturing complementary information among the basic ranking methods. Inspired by this strategy, SMI-BLAST [19] proposes an improved PSSM with lower false-positive rates by combining the LTR and the PSI-BLAST.
The deep neural networks (DNN) are suitable for learning characteristics from protein sequence, whose derived protein features are applicable for different downstream tasks [28]. UDSMProt [29] is a DNN-based language model combined with a pretrain strategy, detecting the remote homology relationships by building a classification model. ProtTrans [30] selects six well-known language models (T5 [31], Electra [32], BERT [33], Albert [34], Transformer-XL [35] and XLNet [36]) and applies them to various classification tasks, such as subcellular localization [37]. The experimental results also prove that these language models capture the global constraints of protein structures after model tuning.
Although the language model based on deep neural networks with a pretrained strategy is able to effectively learn the protein features, these features still lack biological information for remote homology detection. The reason is that the protein features derived from the pretrained language model may cover too complicated information to be used for remote homology relationship, such as structural information. For example, the performance of UDSMProt (with the pretrained language model) is lower than that of ProtDec-LSTM [38] (without the pretrained language model). The reason is that the protein features extracted by ProtDec-LSTM contains more useful information to detect proteins with remote homology. As a result, we should study a novel language model that learns protein features covering multiple protein properties.
The similarities between protein sequences and sentences can be summarized at four levels: (i) symbol level, protein sequences and sentences are composed of a series of symbols; (ii) they follow a certain grammatical rule; (iii) protein sequence and sentences have hierarchical structure; (iv) both protein sequences and sentences convey meaning, such as function or semantics. To achieve accurate protein remote homology detection, a new language model must utilize multi-level information to learn high-level semantic relationships.
The protein cubic language model (PCLM) is a powerful method for learning protein representations from multiple protein properties and has been successfully applied to identify protein disorder molecular functions [39]. Central to PCLM is the notion of representing the general information associated with proteins as a cube, where each face or slice embodies a distinct type of characteristic information. This concept is particularly useful for remote homology detection, as it accounts for various factors influencing the relationships among proteins, such as structural information.
In our proposed computational predictor, PreHom-PCLM, we employ motif-based CNNs (MotifCNNs) to extract features from structural motifs and assemble them into a motif feature cube, which is then input into the PCLM. The PCLM, which combines 2D convolution neural network (2D CNN) and transformer layers (TRM), captures latent remote homology relationships among proteins more effectively than traditional methods by learning from the multi-level information provided by the motif feature cube. Our experimental results demonstrate that PreHom-PCLM outperforms competing methods, highlighting the effectiveness of the PCLM. Moreover, the manifold embedding of protein features generated by PreHom-PCLM exhibits a robust discriminative capacity for proteins, ensuring its structural reliability.
METHODS
Detecting the remote homology by PreHom-PCLM includes two steps, building motif feature cube and feeding this cube into the PCLM, to predict the probability over protein superfamily types (Figure 1). The first step extracts motif features covering multi-level information by motif-based neural networks, then these motif features are fed into PCLM to improve the accuracy of detecting remote homology relationships in the second step.
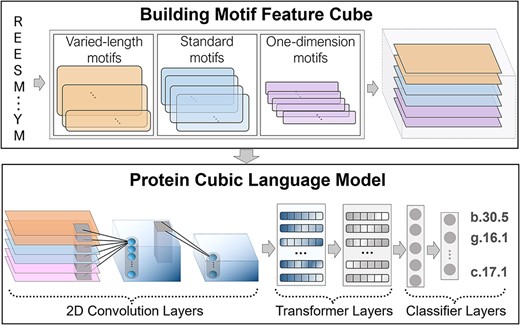
The flowchart of PreHom-PCLM. In the two-step process of PreHom-PCLM, as illustrated in the figure, we first convert the query protein into six types of motif features using a Motif-based CNN, which is built from three groups of motifs. These motif features are then assembled into a motif feature cube. Subsequently, the motif feature cube is input into the PCLM, which consists of 2D CNN and TRM. The PCLM learns latent remote homology relationships among protein sequences and outputs a series of probabilities against superfamily types using a FC-based classifier. Finally, we assign the superfamily type with the highest probability to the query protein as the retrieval results.
Motif convolutional kernels
Proteins are folded by the linear sequence of amino acids, some of which contain unique motifs determining their structures and functions. We introduced the protein motif and build motif feature cube to improve the accuracy of predicting remote homology relationship from protein sequence. The protein sequence information is always provided by the PSSM, the normalized version of which is the position-specific frequency matrix (PSFM). We construct the PSFM by using PSI-BLAST (num_alignments = 1000, evalue = 1e−3, num_iterations = 3) searching against the nrdb90 data set [40]. The PSFM can represented as
where |$\text{PSFM}\in{\mathbb{R}}^{S\times 20}$|, |$S$| is the sequence length and each entry |${p}_{i,j}$| denotes the frequency that the jth amino acid appears in position i of a protein sequence. We design three groups of motifs to extract multi-level information from PSFMs. They are the varied-length motif, standard motif and 1D motif, where each group includes 128 motifs selected from the MegaMotifBase [41, 42]. They are formalized as
where |$l$| denotes the size of each motif. The varied-length motifs keep the same size as the original structural motifs. The standard motifs share an exact size of 7, which is suitable for learning the local sequence pattern. The 1D motifs share an exact size of 1, designed for learning the local position information. These motifs are used for the convolutional kernel of 1D convolution neural network (1D CNN) layers in the MotifCNN. Their weights are all initialized randomly rather than the original ones of structural motifs. We design three styles of MotifCNNs against the varied-length, standard and 1D groups of motifs. Each MotifCNN generates the motif features as
where |${\text{F}}_{motif}\in{\mathbb{R}}^{S\times k}$|, k = 128, denoting the number of motifs in each group, and each element |${f}_{i,j}$| describes the relationship between motif and protein sequence.
In addition, we add a 1D maximum-pooling and average-pooling layer against each MotifCNN to obtain two different motif features, |${\text{F}}_{2i-1},{\text{F}}_{2i}\in{\mathbb{R}}^{H_p\times k},i\in \left\{1,2,3\right\}$|. We finally get six styles of motif features from three groups of motifs.
The architecture of PCLM
The Protein Language Model (PLM) is an effective way for protein sequence analysis [43, 44]. Recent research has proposed the Protein Cubic Language Model (PCLM) to the identification of protein disordered molecular function [39]. The core concept of the PCLM is to fuse multiple characteristics for a specific task. Therefore, this concept is suitable for protein remote homology.
In order to capture the patterns of the superfamilies, we extend the PCLM [39] by incorporating the structural motifs, and proposed a predictor called PreHom-PCLM for protein remote homology detection. The details of PCLM will be introduced in the following.
A motif feature cube |${\text{F}}_{cube}\in{\mathbb{R}}^{6\times{H}_p\times k}$| based on structural motifs is formalized as
where |${\text{F}}_j\in{\mathbb{R}}^{H_p\times k,},j\in \left\{1,2,\dots, 6\right\}$| denotes the jth style of feature extracted by the MotifCNN. This motif feature cube contains information from different levels. For example, the motif features |${\text{F}}_1$| and |${\text{F}}_2$| extracted by MotifCNN with varied-length motifs may include structural information. This motif feature cube is then fed into the PreHom-PCLM for protein remote homology detection. The PreHom-PCLM is formalized as
where the |$\left[{\text{F}}_1;{\text{F}}_2;\dots; {\text{F}}_j;\dots \right]$| denotes a feature cube composed of multi-level features. The |${g}_L\cdotp \dots \cdotp{g}_l\cdotp \dots \cdotp{g}_2\cdotp{g}_1$| represent L feature mapping functions, such as linear or non-linear mapping function.
2D convolutional layers for motif feature combination
We first transpose the motif feature cube |${\text{F}}_{cube}$| into |${\text{D}}_a\in{\mathbb{R}}^{H_p\times k\times 6}$| so that the first module in PreHom-PCLM fully learns the multi-level information. The first module is the 2D CNN to capture the relationships among different motif features in the convolution calculating process. The 2D CNN includes two convolution stages (stage1: 128 filter size, |$3\times 3$| kernel size, |$2\times 2$| stride size; stage2: 64 filter size, |$3\times 3$| kernel size, |$1\times 1$| stride size) combined with the batch normalization (BN) technique [45] and Gaussian error linear units (GELU) [46] activation function. At the tail of the 2D CNN, we add a 2D average-pooling layer (|$5\times 5$| kernel size, |$2\times 2$| stride size) to downsample the output of the 2D CNN. After the above steps, we generate a different motif feature cube, |${\text{D}}_b\in{\mathbb{R}}^{H_m\times{W}_m\times{C}_m}$|, where each value measures the relationships of multiple motif features.
TRM for cubic motif feature embedding
The motif feature cube obtained by the 2D CNN in the previous step represents the relationships among motif features. We add a TRM to capture the global association in the motif feature cube. We first transpose the motif feature cube into a 1D embedding |${\text{T}}_a\in{\mathbb{R}}^{C_m\times{E}_m}$|, |${E}_m={H}_m\times{W}_m$|. Then, we introduce a 1D CNN (64 filter size and 1 kernel size) with the BN technique and GELU activation function to process this embedding, and get the output, |${\text{T}}_b\in{\mathbb{R}}^{C_t\times{E}_t}$|, |${C}_t={C}_m$| and |${E}_t={E}_m$|. After that, we feed |${\text{T}}_b$| into the TRM (0.3 dropout rate, 512 hidden sizes of self-attention block). It outputs an embedding |${\text{T}}_c\in{\mathbb{R}}^{C_t\times{E}_t}$|, including the global association learned by the TRM.
Superfamily prediction
We build a multi-classification model for protein remote homology detection, assigning a specific superfamily type for a query protein. We add a classifier with three fully connected (FC) feed-forward layers (16 384, 4096 and 2048 neurons), where each layer combines the BN technique and GELU activation function (no bias). We first change the embedding results |${\text{T}}_c$| into |$x\in{\mathbb{R}}^{C_f},{C}_f={C}_t\times{E}_t$|, and then feed it into this classifier. We add a SoftMax function to convert the classifier’s output scores into a series of probabilities |${p}_k$| against superfamily types. To train this model, we consider cross-entropy as a loss function:
where |${y}_k$| denotes the real label of superfamily |$k$|, |${p}_{\text{k}}$| is the probability of it and |$K$| is the amount of them. For the other detailed model parameter settings, please refer to Table S1.
Training details
The TensorFlow framework (v1.10.0) is employed to implement the PreHom-PCLM. In this study, we build the PreHom-PCLM with a supervised-based training strategy. We design a two-stage training strategy to obtain an optimal model. In detail, we initialize the model parameters in the second stage by the after-trained one in the first stage and select the Adam [47] algorithm (default parameters) to optimize the PreHom-PCLM parameters during the training process. For the detailed training parameters in different experiments, please refer to Table S2.
DISCUSSION
Data sets
We evaluated the performance of our PreHom-PCLM model for remote homology detection on the Structure Classification of Proteins database (SCOP1.75, 0.95 sequence identity) [48] and Structure Classification of Proteins extended database (SCOPe2.07, 0.95 sequence identity) [3]. SCOP1.75 is for constructing training and validation sets, and SCOPe2.07 is for building test and independent test sets. As for SCOP1.75, we first divided it into 10 parts of equal size, one of them as the validation set and the remaining as the training set. We also used the following tools to reduce the redundancy between the two sets: CD-HIT (0.95 sequence identity) [28, 49], MMseqs (0.95 sequence identity and 10e−4 evalue) [16, 28] and PSI-BLAST (10e−4 evalue) [10, 28]. Finally, the training set contains 14 328 proteins covering 1854 superfamilies. The validation set contains 1186 proteins covering 253 superfamilies.
For the test set, we first removed the proteins in the SCOP1.75 data set from the SCOPe2.07 data set and reduced the sequence identity to 0.95 by using CD-HIT [28]. We divided the test set into five parts with an equal number of samples to perform a 5-fold cross-validation. Therefore, the test set contains 13 557 proteins covering 1126 superfamilies.
For the independent test set, we retrieved the proteins that were added to the SCOPe database between 2020–07 and 2021–12 and reduced redundancy by MMseqs (0.95 sequence identity and 10e−4 evalue) [28]. As a result, the independent test set contains 5990 proteins covering 839 superfamilies, none of which overlap either the test set or the training set [50].
Experiments and evaluation metrics
Two experiments were conducted to demonstrate the effectiveness of the PreHom-PCLM: 5-fold cross-validation on the test set and an independent test. We trained a basic model in the training set, which was then tuned for the 5-fold cross-validation and independent test, respectively. In the 5-fold cross-validation, the test set is divided into five equal parts, and one part is used for testing while the remaining four parts are used to tune the base model. In the independent test, the base model is fine-tuned on the entire test set, and the performance is evaluated on the independent test set.
The PreHom-PCLM detects the remote homology relationship by predicting probability distribution to superfamily types and assigns the highest-probability type to the query protein. We used two metrics to measure the performance of different methods. These metrics are ROC1 and ROC50 [51–53].
Ablation study
There are two essential components in PreHom-PCLM to utilize the motif features for detecting protein remote homology relationships, including 2D CNN and TRM. We performed an ablation experiment to study the roles of different components in PreHom-PCLM (Table 1). The PreHom-PCLM without the two components achieves the lowest performance, indicating that these two components are essential. This is because the two components contribute to capturing the relationships among the motif features. In detail, the 2D CNN learns the local relationship of different motif features; the TRM strengthens this relationship by self-attention mechanisms. Furthermore, the 2D CNN is more important than TRM because the performance of PreHom-PCLM decreases more obviously after removing the 2D CNN compared with the PreHom-PCLM without using TRM. The reason is that the protein features learned by 2D CNN are the foundation for the TRM to learn more complicated information. This result in Table 1 is not surprising because the two components in PreHom-PCLM capture different relationships among the motif features contributing to the accuracy improvement of remote homology detection.
The performance of PreHom-PCLM predictors based on different components and their combinations
| Method | 2D CNN | TRM | ROC1 | ROC50 |
|---|---|---|---|---|
| PreHom-PCLM | ✔ | ✔ | 0.9857 | 0.9937 |
| ✔ | ✗ | 0.9820 | 0.9926 | |
| ✗ | ✔ | 0.9538 | 0.9797 | |
| ✗ | ✗ | 0.9453 | 0.9770 |
| Method | 2D CNN | TRM | ROC1 | ROC50 |
|---|---|---|---|---|
| PreHom-PCLM | ✔ | ✔ | 0.9857 | 0.9937 |
| ✔ | ✗ | 0.9820 | 0.9926 | |
| ✗ | ✔ | 0.9538 | 0.9797 | |
| ✗ | ✗ | 0.9453 | 0.9770 |
The performance of PreHom-PCLM predictors based on different components and their combinations
| Method | 2D CNN | TRM | ROC1 | ROC50 |
|---|---|---|---|---|
| PreHom-PCLM | ✔ | ✔ | 0.9857 | 0.9937 |
| ✔ | ✗ | 0.9820 | 0.9926 | |
| ✗ | ✔ | 0.9538 | 0.9797 | |
| ✗ | ✗ | 0.9453 | 0.9770 |
| Method | 2D CNN | TRM | ROC1 | ROC50 |
|---|---|---|---|---|
| PreHom-PCLM | ✔ | ✔ | 0.9857 | 0.9937 |
| ✔ | ✗ | 0.9820 | 0.9926 | |
| ✗ | ✔ | 0.9538 | 0.9797 | |
| ✗ | ✗ | 0.9453 | 0.9770 |
Five-fold cross-validation
We benchmarked PreHom-PCLM against three alignment methods (HHblits [5, 15], HMMER [13, 14] and PSI-BLAST [10, 11]), ProtDec-LTR 3.0 [26] and PL-HHblits, PL-HMMER and PL-BLAST [23] (Table 2).
The comparison results on the test set
| Method | ROC1 | ROC50 |
|---|---|---|
| PreHom-PCLM | 0.9356 | 0.9618 |
| PL-HHblitsa | 0.9207 | 0.9426 |
| PL-HMMERa | 0.9043 | 0.9188 |
| PL-BLASTa | 0.9047 | 0.9155 |
| ProtDec-LTR 3.0b | 0.9002 | 0.9016 |
The comparison results on the test set
| Method | ROC1 | ROC50 |
|---|---|---|
| PreHom-PCLM | 0.9356 | 0.9618 |
| PL-HHblitsa | 0.9207 | 0.9426 |
| PL-HMMERa | 0.9043 | 0.9188 |
| PL-BLASTa | 0.9047 | 0.9155 |
| ProtDec-LTR 3.0b | 0.9002 | 0.9016 |
These alignment methods are based on the profile feature, which iteratively search the query sequence against the protein database by their extracted sequence profile feature. A profile feature like the HMM profile is a probability matrix that describes the probability of the amino acid occurring in different positions of a sequence. However, the profile feature contains nonhomologous errors, leading to prediction biases.
The ProtDec-LTR 3.0 applied an ensemble and protein network methods into fusing alignment and profile features-based methods, which reduce some of the nonhomologous errors. The PL-search method builds a double-link database to prevent the nonhomologous proteins from the alignment results. It also utilizes the profile-link database and the Jaccard measurement strategy to expand the number of remote homology sequences.
However, PL-search is not completely effective in preventing nonhomologous errors since they rely on alignment methods. Experimental evidence suggests a decrease in the performance of PL-HHblits (ROC1: 0.8277, ROC50: 0.8849) when the extent of its sequence retrieval matches the size of PreHom-PCLM’s training data. This is attributable to the sequence alignment method’s limited capacity, which measures only the similarity between sequences without considering the structural similarity of proteins. Such limitations can engender nonhomologous errors, misleading the ranking optimization algorithm of PL-search when retrieval data are insufficient. As a solution for enhancing protein remote homology detection, we introduce PreHom-PCLM, a method that fuses different features. Our strategy includes three types of neural networks based on motifs, designed to extract structural information as well as other pertinent information such as local sequence patterns and positional information. PreHom-PCLM surpasses PL-HHblits in performance, thus demonstrating its effectiveness in integrating multiple motif features and addressing more nonhomologous errors than PL-search.
Independent test
Despite the improved performance of our PreHom-PCLM, the generalization ability has yet to be evaluated. To evaluate the generalization ability of the PreHom-PCLM and PL-search methods, we conducted an independent test (Table 3). The PL-search performs better on the independent test set than on the test set because the PL-search eliminates nonhomologous proteins. However, it is difficult to improve the PL-search anymore because of the limited quality of the alignment methods. The PreHom-PCLM avoids this problem by automatically fusing multi-level information by using the three styles of motif-based neural networks.
PreHom-PCLM outperforms the PL-search for independent test
| Method | ROC1 | ROC50 |
|---|---|---|
| PreHom-PCLM | 0.9857 | 0.9937 |
| PL-HHblitsa | 0.9749 | 0.9777 |
| PL-HMMERa | 0.9340 | 0.9370 |
| PL-BLASTa | 0.9405 | 0.9707 |
| Method | ROC1 | ROC50 |
|---|---|---|
| PreHom-PCLM | 0.9857 | 0.9937 |
| PL-HHblitsa | 0.9749 | 0.9777 |
| PL-HMMERa | 0.9340 | 0.9370 |
| PL-BLASTa | 0.9405 | 0.9707 |
aReproduced the PL-search for independent test.
PreHom-PCLM outperforms the PL-search for independent test
| Method | ROC1 | ROC50 |
|---|---|---|
| PreHom-PCLM | 0.9857 | 0.9937 |
| PL-HHblitsa | 0.9749 | 0.9777 |
| PL-HMMERa | 0.9340 | 0.9370 |
| PL-BLASTa | 0.9405 | 0.9707 |
| Method | ROC1 | ROC50 |
|---|---|---|
| PreHom-PCLM | 0.9857 | 0.9937 |
| PL-HHblitsa | 0.9749 | 0.9777 |
| PL-HMMERa | 0.9340 | 0.9370 |
| PL-BLASTa | 0.9405 | 0.9707 |
aReproduced the PL-search for independent test.
To assess the predictive performance of proteins with low sequence identity, we created a subset of 23 protein sequences by eliminating those from the original independent test set that had a sequence identity > 0.2 using the MMseqs tool. We then re-evaluated PreHom-PCLM and PL-search on this subset and observed that PreHom-PCLM exhibited higher accuracy compared to PL-search. For detailed results, please refer to Table S6.
Furthermore, we evaluated the performance of 3D protein structure prediction using our method. To accomplish this, we utilized Clustal Omega [54, 55] to compute multiple sequence alignment (MSA) results for 23 distinct test sequences that had <20% sequence identity with the training data set. We compared the TM-scores (Template Modeling score) and LDDT scores (Local Distance Difference Test score) of the structure predictions generated by trRosetta [56–59]. These predictions were derived from MSAs generated using PreHom-PCLM, PL-HHblits and HMMER algorithms (Figure 2A–C). The comparison demonstrated that PreHom-PCLM achieves more accurate predictions than PL-HHblits and HMMER. Moreover, when compared with trRosettaX-single [59], a language model-based method that takes a single sequence as input, PreHom-PCLM and trRosettaX-single perform comparably, surpassing PL-HHblits and HMMER. Notably, when focusing on the LDDT metric, emphasizing the accuracy of protein residue-level prediction, PreHom-PCLM’s advantage becomes more pronounced, even surpassing the trRosettaX-single method (Figure 2, Part II).
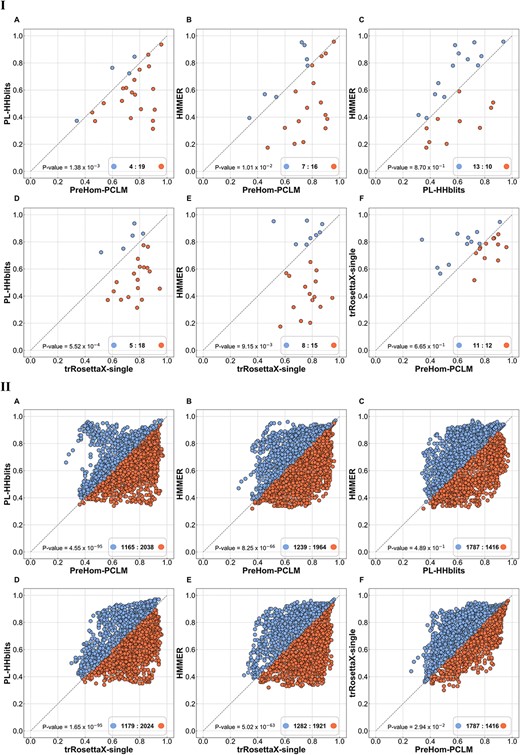
PreHom-PCLM accurately predict structure by using trRosetta. The x- and y-axis denote the predicted TM-score (Part I) or the predicted LDDT score (Part II) of different methods based on trRosetta. Sample points located to the right of the diagonal line on the graph indicate a higher precision in the structure prediction of method in x-axis, whereas points placed on the left demonstrates y-axis method’s higher prediction accuracy. In addition, no PDB template information was used in this protein structure prediction experiment.
According to these results, we identified two main reasons for the effectiveness of our proposed PCLM: (i) we extracted features from structural motif to capture both sequence and structural characteristics of proteins; (ii) we employed the 2D CNN and Transformer components to constructs PCLM so as to facilitate the learning of high-level semantic relationships among proteins that are relevant for remote homology detection.
The improvement of the proposed remote homology detection method on protein structure prediction can be summarized as follows: (i) for proteins that are difficult to align using traditional sequence alignment methods, our method can provide more accurate structural prediction results by generating MSAs using proteins with remote homology relationships; (ii) our method achieves comparable or better prediction results compared to PL-HHblits, without relying on manually constructed sequence retrieval data sets; (iii) PreHom-PCLM performs comparably to trRosettaX-single, which is based on protein language model, indicating that determining remote homologous relationships is useful for predicting protein structure.
Protein structure analysis by AlphaFold2
AlphaFold2, developed by DeepMind, is a cutting-edge deep learning method that has revolutionized the analysis of protein structures. In this study, we utilize the local version of ColabFold (v1.5.1) [60], which allows the use of custom MSAs as input. ColabFold combines the fast homology search capabilities of MMseqs2 with either AlphaFold2 or RoseTTAFold to provide accelerated predictions of protein structures and complexes. In this study, ColabFold employs weights that are aligned with the official AlphaFold2 to ensure the alignment of predictions between ColabFold and official AlphaFold2. To maintain simplicity and clarity, we will use the term ‘AlphaFold2’ to refer to ColabFold, which incorporates the official weights of AlphaFold2. The experimental samples consist of the same 23 proteins from the independent test set mentioned in the previous section. We compare the LDDT score of the structure predicted by AlphaFold2 using different MSAs generated by PreHom-PCLM, PL-HHblits and HMMER (Figure 3, Part I: A–C). In addition, we compare these results with trRosettaX-single (Figure 3, Part I: D–F). Finally, we compare the structure predictions of AlphaFold2 and trRosetta using the same set of MSAs as input based on the metrics of LDDT score (Figure 3, Part II: A–C) and TM-score (Figure 3, Part II: D–F).
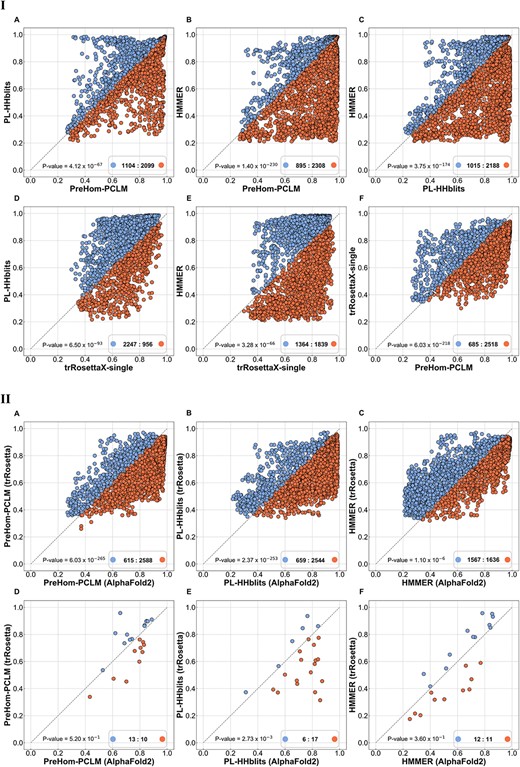
PreHom-PCLM accurately predict structure by using AlphaFold2. The x- and y-axis denote the predicted LDDT score (Part I and Part II: A–C) or TM-score (Part II: D–F) of different methods based on AlphaFold2. Additional details are consistent with Figure 2.
Based on Figure 3, the following conclusions can be drawn: (i) AlphaFold2 attained superior accuracy by employing MSAs generated by PreHom-PCLM, surpassing the performance of trRosettaX-single; (ii) AlphaFold2 demonstrated superior performance compared to trRosetta, especially regarding the LDDT score, a metric quantifying accuracy at the protein residue level.
The PCLM extracts more available information from motif features
The remote homology relationships depend on multiple protein properties, such as structural information and local sequence patterns. In this study, three motif-based neural networks were constructed in order to get different types of motif features that covered various protein properties. To confirm the effectiveness of PCLM, we compare the PreHom-PCLM with the other three methods (1D CNN, AvgPool and MaxPool), where the motif features are linearly concatenated, and fed to the 1D CNN, average-pooling and maximum-pooling layers, respectively. In contrast, the PreHom-PCLM builds a motif feature cube, and feeds it into the PCLM composed of 2D CNN and TRM so as to gain a deeper insight of the potential remote homology relationship.
Table 4 demonstrates that PreHom-PCLM outperforms the other methods, indicating the following: (i) constructing a motif feature cube and feeding it into the PCLM is more suitable than the other feature fusing strategies; (ii) the CNN architecture helps to learn the differences among the local patterns of motif features for remote homology detection; (iii) the 2D CNN employed in the PCLM is better than 1D CNN because it learns more available correlation patterns of motif features for detecting protein remote homology relationships.
The results of different approaches for using the motif features on independent test set
aLinearly concatenate the motif features.
The results of different approaches for using the motif features on independent test set
aLinearly concatenate the motif features.
PreHom-PCLM captures the global structural constraint in proteins
The remote homology relationships are related to the structure-level information [3]. The structural class is the top hierarchical organization in the SCOP/SCOPe databases. The PreHom-PCLM learns the remote homology relationships by integrating multiple motif features covering multi-level information, like structural information. We plot the protein distribution by the manifold learning algorithm to compare the raw sequence and the protein feature distributions to show that the PreHom-PCLM has captured the structural information (Figure 4). As shown in Figure 4A, the structure classes are hard to distinguish based on the raw sequence distributions. Figure 4B shows that even the PreHom-PCLM maps the proteins into the same high-dimensional feature space, but they lack discriminative power at intra-class and inter-class levels without training. Figure 4C shows that the features extracted by trained PreHom-PCLM are highly discriminative, aggregating the proteins of the same structural class into the same cluster and distinguishing these clusters from each other.
![The feature distributions of proteins in the independent test set. We plot a SCOPe structural class as a point in these figures. (A) shows the original distributions based on the Edit Distance [61]. (B) visualizes the random representation based on the untrained PreHom-PCLM. (C) visualizes the final representation of the trained PreHom-PCLM. We employ the t-SNE algorithm (perplexity = 100, learning_rate = ‘auto’, init = ‘pca’, random_state = 0, metric = ‘precomputed’ in (A) and ‘euclidean’ in (B) and (C)) to convert the high-dimensional protein representation into 2D position distribution.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/bib/24/6/10.1093_bib_bbad347/2/m_bbad347f4.jpeg?Expires=1749540859&Signature=f1cjs49fe9luL21xm~h5x0xI-fpioDx0kty9-8UESghmwKvdDEy~6Cvac5B9Ho3Ct5uRF9mMG4KGZF6k-FND0DpuyA0Q7klrZB8XtjVuBo6B714q3jcHwsDP9S9eQ6rYVAUvXfgeRDL~D57F8roHISZBltxEUX8ns02ebphwZX9Ul0UsPD-7e5gA1LvhqVc3bT6bTsUB3PO4Em-U2mCmKrb8kPZpGENBH0uxZPlkjGOwPS02VDzsAvl7tdeyDFQXPqUblQ6xQCYrpfob-wPBeETvuDZOTm1FLdFv~YfTJkxcnpLdqpDobw3kUzcWmTraXUsGTsFbQdP4SCiGnZDS~A__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
The feature distributions of proteins in the independent test set. We plot a SCOPe structural class as a point in these figures. (A) shows the original distributions based on the Edit Distance [61]. (B) visualizes the random representation based on the untrained PreHom-PCLM. (C) visualizes the final representation of the trained PreHom-PCLM. We employ the t-SNE algorithm (perplexity = 100, learning_rate = ‘auto’, init = ‘pca’, random_state = 0, metric = ‘precomputed’ in (A) and ‘euclidean’ in (B) and (C)) to convert the high-dimensional protein representation into 2D position distribution.
CONCLUSION
We employed the Protein Cubic Language Model (PCLM) to learn protein representation based on multiple motif features and proposed a predictor called PreHom-PCLM for protein remote homology detection. Experimental results show that PreHom-PCLM outperforms the other state-of-the-art predictors, which is a promising approach to extract and fuse different patterns of proteins (e.g. structure, sequence pattern). Moreover, the PreHom-PCLM does not rely on a large-scale MSA, which reduces computational cost and avoids potential errors introduced by alignment.
The principles and techniques of the PCLM, inspired by natural language processing, offer potential advancements in protein structure and function analysis. Specifically, remote homology relationships among proteins underpin the diversity of protein structures in various organisms. By learning characteristics at sequence, structure and function levels from PreHom-PCLM outputs, the accuracy of identifying intrinsically disordered regions can be improved [39]. Moreover, PreHom-PCLM can predict remote homology relationships for small-sized proteins (<50 amino acids), such as peptides that lack alignment results [62]. This addresses the issue of low accuracy in peptide type identification caused by insufficient evolutionary information. In future research, we plan to further explore protein language models like PCLM to advance studies in predicting protein function, like Gene Ontology terms.
PreHom-PCLM employed a novel language model (protein cubic language model, PCLM) for protein remote homology detection. It treats different protein properties related to the distant homology as different faces of a cube and incorporates them by the PCLM.
PreHom-PCLM developed different feature extractors based on protein motifs to construct PCLM so as to cover multiple characteristic information related to remote homology relationships.
The benchmark and independent test set results suggested that PreHom-PCLM outperforms the other competing methods. The free-access web server is located at http://bliulab.net/PreHom-PCLM.
FUNDING
The National Natural Science Foundation of China (Nos 62271049, U22A2039, 62250028, 62102030), Beijing Institute of Technology Research and Innovation Promoting Project (Grant No. 2022YCXZ008).
Author Biographies
Jiangyi Shao is a PhD candidate at the School of Computer Science and Technology, Beijing Institute of Technology, China. His expertise is in bioinformatics.
Qi Zhang is a master student at the School of Computer Science and Technology, Beijing Institute of Technology, China. His expertise is in bioinformatics.
Ke Yan, PhD, is an assistant professor at the School of Computer Science and Technology, Beijing Institute of Technology, Beijing, China. His expertise is in bioinformatics, natural language processing and machine learning.
Bin Liu, PhD, is a professor at the School of Computer Science and Technology, Beijing Institute of Technology, Beijing, China. His expertise is in bioinformatics, natural language processing and machine learning.
References
Sergey I, Christian S. Batch normalization: accelerating deep network training by reducing internal covariate shift. In: Francis B, David B. (eds),

{kind=link}
{kind=link}
{kind=link}
{kind=link}