




Abstract
Covalent inhibitors have received extensive attentions in the past few decades because of their long residence time, high binding efficiency and strong selectivity. Therefore, it is valuable to develop computational tools like molecular docking for modeling of covalent protein–ligand interactions or screening of potential covalent drugs. Meeting the needs, we have proposed HCovDock, an efficient docking algorithm for covalent protein–ligand interactions by integrating a ligand sampling method of incremental construction and a scoring function with covalent bond-based energy. Tested on a benchmark containing 207 diverse protein–ligand complexes, HCovDock exhibits a significantly better performance than seven other state-of-the-art covalent docking programs (AutoDock, Cov_DOX, CovDock, FITTED, GOLD, ICM-Pro and MOE). With the criterion of ligand root-mean-squared distance < 2.0 Å, HCovDock obtains a high success rate of 70.5% and 93.2% in reproducing experimentally observed structures for top 1 and top 10 predictions. In addition, HCovDock is also validated in virtual screening against 10 receptors of three proteins. HCovDock is computationally efficient and the average running time for docking a ligand is only 5 min with as fast as 1 sec for ligands with one rotatable bond and about 18 min for ligands with 23 rotational bonds. HCovDock can be freely assessed at http://huanglab.phys.hust.edu.cn/hcovdock/.
Introduction
Covalent protein–ligand interactions play an important role in drug discovery [1–6]. Covalent inhibitors generally refer to the molecules in which the electrophilic part of the ligand forms a covalent bond with the nucleophilic residue on the receptor. Covalent inhibitors bind to their protein targets through both covalent and non-covalent interactions, and therefore exhibit a higher potency, practically long residence time and higher binding affinity than traditional non-covalent inhibitors [7–10]. Over 50 covalent modified drugs have been approved by the FDA [11]. Among them, drugs with cysteine as the target reaction residue account for one third of them [12]. For example, Pfizer has launched an oral covalent drug [13], which illustrates the pivotal position and importance of covalent drugs based on cysteine as the reactive residue.
Given the high cost and technical difficulties of experimental methods, computational tools like molecular docking have played an increasing role in the drug discovery process [14–27]. Correspondingly, a number of approaches for covalent ligand docking and virtual screening have been developed, including WIDOCK [28, 29], AutoDock [30], CovalentDock [31], CovDock [32, 33], Cov_DOX [34], DOCKovalent [35], DOCKTITE [36], FITTED [37–40], GOLD [41–43], MOE [44] and ICM-Pro [45, 46]. WIDOCK is an AutoDock4-based virtual screening protocol that supports covalent ligand docking by incorporating ligand reactivity to cysteine residues [28, 29]. AutoDock implements a flexible side chain method to model the covalent protein–ligand interactions [30]. In the flexible side chain method, a ligand coordinate file is modified by connecting two target residue atoms, C|$_{\beta }$|-SG in the case of cysteine or at the site of alkylation. CovDock is a covalent docking protocol adapted from Glide in the Schrödinger suite [32, 33]. It mutates the reactive residue at the binding site to alanine, which allows the warhead to get closer to the target residue and avoids adverse clashes. After that, CovDock employs the ConfGen [47] to generate the ligand conformations for the docking calculation, and retains up to 10 ligand poses. In the following steps, the reactive residue is mutated back, and the rotamer states of reactive residue are sampled to form a covalent bond with the top-scoring ligand-binding conformation based on geometric criteria. Cov_DOX is a hybrid method for covalent docking, which involves quantum mechanical (QM) calculations for covalent interactions [34]. There are three docking/scoring protocols for Cov_DOX, of which the medium level PM7 [48] adopts a semi-empirical scoring method and has the best balance between speed and accuracy of covalent docking. DOCKovalent is a covalent docking software based on DOCK3.6 [49], which constrains the warhead of the ligand close to the reactive residue with predefined distance, angle and dihedral to reduce the sampling conformation space. DOCKTITE is a covalent docking workflow based on MOE [44], which attaches the warhead to the reactive residue via a pseudo receptor side chain connection [36]. FITTED is a docking tool integrated into the FORECASTER Platform. It starts with non-covalent docking. If the warhead is located at the neighboring region of reactive residue, a covalent bond will be formed [37–39]. GOLD is a genetic algorithm-based docking program. Its covalent implementation is based on the overlap of “link-atom” in both the ligand and reactive residue of the protein [41–43]. ICM-Pro is developed by Molsoft LLC, which includes a protocol for covalent docking [45, 46]. Given the type of reaction, ICM-Pro covalently attaches all stereoisomers resulting from the addition of electrophilic warheads to cysteine side chains, automatically converting the pre-reaction ligands into ‘pseudo-ligands.’
Traditionally, covalent inhibitors are discovered by adding electrophiles to known reversible inhibitors that suitably bind to a reactive residue (cysteine) [50–53]. More recently, covalent inhibitors are also being designed by screening of covalent fragment libraries [54–59]. However, no matter which docking method is adopted, the balance of the contribution of the covalent part and the non-covalent part to the interaction energy has always been a difficult issue for covalent binding [60, 61]. Addressing this issue, we have developed a new covalent docking algorithm, named HCovDock, for covalent protein–ligand binding. HCovDock adopts a search strategy of step-by-step construction, firstly sampling the covalent part exhaustively, and then gradually searching the conformational space of the subsequent non-covalent parts like a long branch step by step. To balance the contributions between covalent and non-covalent interactions, we have modified the non-bonded energy terms of the AMBER force field [62] to describe the intermolecular interaction of the ligand with the protein. HCovDock is extensively evaluated on a benchmark of diverse protein–ligand complexes and compared with state-of-the-art docking methods. The virtual screening power of HCovDock is also evaluated on a public benchmark of 10 receptors. It shows that our docking algorithm significantly improved the docking accuracy while maintaining the high computational efficiency.
Materials and methods
Structure preparation
The first step of the HCovDock is to prepare the receptor and ligand structures for docking input. Specifically, the protein is separated from its covalent ligand. Metal ions, cofactors and structural water molecules are also removed. The final protein receptor is saved in MOL2 format with hydrogens and atomic charges added. For ligands, to reduce the impact of the native conformation, we use the Conformator [63] and OpenBabel [64] to generate five conformations each from the ligand pdb file, yielding a total of 10 conformations for each ligand. The generated ligand conformations are added with hydrogens and charges using the Gasteiger method in UCSF Chimera [65].
Pose sampling
The binding conformations of a ligand in HCovDock are sampled using an incremental construction method modified from UCSF DOCK 4 [66], in which two virtual atoms are added to the warhead for the ligand attached to the receptor by superimposing two virtual atoms of the ligand to the reactive residue of the receptor (Figure 1A). Prior to the sampling step, several preparatory steps are used to divide the ligand into different rigid segments. First, we take the warhead as the center and identify the rotatable bonds of the ligand (Figure 1B). Second, we gradually divide the whole small molecule into several rigid segments from the inside to the outside, according to the bonding relationship of acyclic rotatable bonds (Figure 1C). The last step is to arrange the rigid fragments into a tree structure starting from the warhead. The atom adjacent to each rotatable bond is transferred to the inner segment since it is not affected by the rotation of the bond. We define an id for each layer, where the warhead is located in layer 0 with a larger id for the outside layer (Figure 1D).
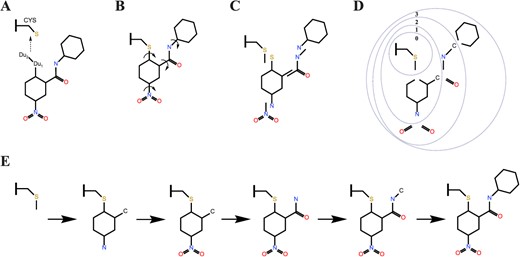
A scheme of binding conformation of ligand sampling. (A) Attached ligand to the receptor. (B) Identification of rotatable bonds. (C) Divided small molecules into distinct rigid segments. (D) Divided the overlap parts into the inner segment and the id of the layer increases from the inside to the outside. (E) Incremental construction started with the warhead layer (layer 0) and proceeded outward.
The next stage of the conformation search is the sampling of the warhead segment. We set a minimum angular spacing of 2.5 degrees for the dihedral angles of covalent bonds formed between the ligands and receptor, with sampling spaces ranged −180|$^\circ $| to −40|$^\circ $| and from 40|$^\circ $| to 180|$^\circ $| for cysteine. The scope of the sampling space is based on the properties of the alcohol–sulfur structure and the fact that no trans-structure is observed in the experimental structures. Furthermore, we remove the conformations of the warhead segment that have severe steric clash with the receptor. The remaining conformations are saved as the initial structures for subsequent sampling.
Then, the incremental construction of the ligand is performed by starting with the warhead layer and proceeding outward (Figure 1E). Before searching for the outer rigid segments, we obtain the possible torsion angles of rotatable bonds by comparing the torsion to a library of flexible bond definitions [66]. HCovDock rotates the rotatable bond by each of possible angles, generating multiple conformations. During this process, each cycle of conformational search involves adding a rigid segment to the current set of conformations and sampling the appropriate torsion of the rotatable bond. To avoid an exponential increase in the number of sampled conformations, a pruning operation for the conformation set is applied during the incremental construction.
In addition, it is also important to maintain a heterogeneous population of configurations to reduce the chance of falling into a local minimum during the conformational search. To increase the abundance of sampling, we do not prune the conformation sets after the layer 0 and layer 1 of sampling. Instead, we add a bump filter to improve the computational efficiency after the second layer of sampling. Namely, if the total bumps between the protein and the sampled ligand pose is larger than a certain number, the ligand pose will be abandoned. Here, a bump is defined as a pair of protein’–ligand atoms that are within the 0.5 times of the sum of the van der Waals (VDW) radii of two atoms [66]. Finally, all the ligand binding conformations are re-optimized, and clustered with an RMSD cutoff is 0.5 Å. Namely, if two ligand poses are within 0.5 Å, the one with the better score is kept.
It is noted that all the rings in the ligand are treated as rigid fragments during the docking process of HCovDock. Therefore, the flexibility of large rings in macrocyclic molecules cannot be considered during the conformational search of our incremental construction for the ligand. Instead, the flexibility of macrocyclic molecules with large ring substructures may be partly taken into account by docking an ensemble of ligand conformers that are pre-generated by a third-party program like Conformator [63] or OpenBabel [64], as used in this study.
Scoring function
Theoretically, in addition to the non-bonded LJ interaction and Coulombic energy, the bond length and angle terms as well as torsional energy need to be included into the ligand intramolecular energy during the ligand conformational energy. However, for the sake of computational efficiency, we follow the same strategy as that used in the conformational search for DOCK [66]. Namely, the bond lengths and angles are taken from the input ligand conformation and will not change during the conformation search. In addition, torsional rotations are only allowed for the bonds with a low energy barrier. Namely, two torsions are allowed for sp|$^2$|=sp|$^2$| double bonds, three torsions for sp|$^3$|–sp|$^3$| single bonds and six torsions for sp|$^2$|–sp|$^3$| single bonds [66]. As such, given the optimized bond length and angle in the initial ligand conformations and the low torsional energy difference among the sampled conformations, the binding modes is expected to be determined by the non-bonded interactions much more than by the bonded energy.
Another important energy in protein–ligand binding is the hydrogen bonding interaction. However, we do not explicitly consider such interactions in the present study because of three reasons. First, due to its electrostatic interaction nature, the hydrogen bonding interaction can be fairly considered by a combination of VDW interactions and Coulombic energy, as used in DOCK [66]. Second, the calculation of hydrogen bonding interactions strongly depends on the positions of involved hydrogens. This may introduce errors into the calculated hydrogen bonding energy because the positions of hydrogens are not optimized during the docking calculation. Third, the calculation of hydrogen bonding interactions will increase the computational cost and will thus significantly lower the computational efficiency of the docking process.
Optimization
HCovDock employs a SIMPLEX minimization method to optimize the binding score during the on-the-fly search of the ligand binding conformations. Quaternions are used to represent the angles of rotation for ensuring the numerical stability. The inner torsion changes the conformation more than the outer torsion. To maintain the consistency in the optimization process and mitigate the impact of this distortion, we scale the inner torsion step according to the number of the inner torsion and the current layer.
Benchmark data sets
The benchmark for validate HCovDock is taken from the data set of 207 diverse covalent protein–ligand complexes prepared by Scarpino et al., which have been used to evaluate various covalent docking programs [68]. As such, abundant data are available for comparative analysis on this benchmark. The data set is collected from the protein data bank (PDB) [69], and its complexes have a resolution of 2.5 Å or better. It includes 207 unique ligands and 54 targets with different protein classes mainly spanning over proteases (60|$\%$|), kinases (20|$\%$|), GTP (7|$\%$|) and others (13|$\%$|). The number of heavy atoms in the ligand are between 8 and 50, and the atom types are only among the elements of H, C, N, O, F, P, S, Cl and Br. The covalent complexes are annotated based on the seven types of reaction mechanisms occurring between the ligand and the protein: Michael addition, addition to nitrile, aldehyde and ketone, nucleophilic substitution, ring opening, and disulfide bond formation.
To evaluate the screening power of HcovDock, we also use a covalent-ligand virtual screening test set, which has been constructed for CovDock by Zhu et al. [32]. Specifically, Zhu et al. used 10 Michael acceptors to search for acrylamide containing compounds on the Schrodinger internal database and chose the best 100 matches based on fingerprint Tanimoto similarity and property space. The top 10 fingerprint hits for each ligand probe are pooled into a unique set of 44 compounds. The remaining 57 compounds are selected by a property space proximity technique using molecular weight, molar refractivity, lipophilicity of a molecule expressed as log, rotatable bond, hydrogen bond donor, hydrogen bond acceptor, polar surface area and a number of heavy atom descriptors. Finally, a set of 10 acrylamide Michael acceptors is selected as the true binders. In this test set, each acceptor contains 10 active binders and 100 decoy ligands.
Evaluation criteria
The quality of a predicted protein–ligand binding mode is measured by the closeness between the predicted binding mode and the native structure. If a binding mode has an RMSD of heavy atoms < 2.0 Å, it is defined as a successful prediction (or a hit). For evaluation on the benchmark, the docking performance is measured by the success rate, i.e. the percentage of the cases with at least one hit when a certain number of top predictions are considered. During the evaluation, the top-ranked pose is considered by default, though the top 10 poses are also included for comparison with the corresponding results from the literature [34].
Results and discussion
Performance on the benchmark of 207 complexes
Figure 2 shows the success rates of HCovDock in binding mode prediction under the criteria of RMSD < 1.0 Å and <2.0 Åon the benchmark of 207 covalent protein–ligand complexes when the top predictions are considered [68]. The detailed docking results are listed in Supplementary Table S1. For comparison, the figure also gives the corresponding success rates of seven other state-of-the-art covalent-ligand docking programs (Cov_DOX, ICM-Pro, CovDock, AutoDock, GOLD, FITTED and MOE) from the literature [34], where CovDock is a covalent docking protocol adapted from Glide in the Schrödinger suite.
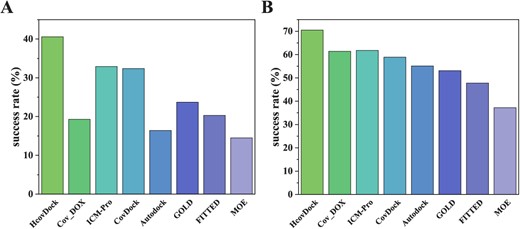
Comparison of the success rates by HCovDock and the other seven docking programs (Cov_DOX, ICM-Pro, CovDock, Autodock, GOLD, FITTED and MOE) on the benchmark of 207 protein–ligand complexes under the criteria of RMSD < 1.0 Å(A) and < 2.0 Å(B) when the top binding mode is considered.
It can be seen from Figure 2 that HCovDock obtains the best performance among the eight docking programs. Specifically, HCovDock obtains the success rates of 40.6% and 70.5% under RMSD <1.0 Å and <2.0 Å, respectively, compared with 19.3% and 61.4% for Cov_DOX(PM7), 32.9% and 61.8% for ICM-Pro, 32.4% and 58.9% for CovDock, while MOE has the least satisfactory performance with 14.5% and 37.2% (Table 1). Another notable in Figure 2 is that HCovDock achieves the consistently highest success rates under both RMSD < 1.0 Å and <2.0 Å, while some other docking methods are criterion-dependent. For example, Cov_DOX and AutoDock obtain a relatively good performance in predicting the ligand poses with RMSD < 2.0 Å, but have significantly lower success rates than other docking approaches in reproducing the ligand binding poses with RMSD < 1.0 Å.
Comparison of the success rates of HCovDock and seven other docking methods under two different RMSD criteria when the top 1 and 10 binding modes are considered, respectively, where the results of the methods other than HCovDock are taken from the literature [34]. The number in bold fonts indicates the highest success rate in the category.
| Type | RMSD | Success rates | |||||||
|---|---|---|---|---|---|---|---|---|---|
| HCovDock | Cov_DOX | ICM-Pro | CovDock | Autodock | GOLD | FITTED | MOE | ||
| Top1 | < 1 Å | 40.6% | 19.8% | 32.9% | 32.4% | 16.4% | 23.7% | 20.3% | 14.5% |
| < 2 Å | 70.5% | 61.4% | 61.8% | 58.9% | 55.1% | 53.1% | 47.8% | 37.2% | |
| Top10 | < 1 Å | 61.8% | 37.7% | 42.0% | 49.8% | 29.5% | 37.2% | 39.6% | 22.7% |
| < 2 Å | 93.2% | 92.8% | 88.0% | 74.4% | 75.4% | 65.2% | 69.6% | 50.7% | |
| Type | RMSD | Success rates | |||||||
|---|---|---|---|---|---|---|---|---|---|
| HCovDock | Cov_DOX | ICM-Pro | CovDock | Autodock | GOLD | FITTED | MOE | ||
| Top1 | < 1 Å | 40.6% | 19.8% | 32.9% | 32.4% | 16.4% | 23.7% | 20.3% | 14.5% |
| < 2 Å | 70.5% | 61.4% | 61.8% | 58.9% | 55.1% | 53.1% | 47.8% | 37.2% | |
| Top10 | < 1 Å | 61.8% | 37.7% | 42.0% | 49.8% | 29.5% | 37.2% | 39.6% | 22.7% |
| < 2 Å | 93.2% | 92.8% | 88.0% | 74.4% | 75.4% | 65.2% | 69.6% | 50.7% | |
Comparison of the success rates of HCovDock and seven other docking methods under two different RMSD criteria when the top 1 and 10 binding modes are considered, respectively, where the results of the methods other than HCovDock are taken from the literature [34]. The number in bold fonts indicates the highest success rate in the category.
| Type | RMSD | Success rates | |||||||
|---|---|---|---|---|---|---|---|---|---|
| HCovDock | Cov_DOX | ICM-Pro | CovDock | Autodock | GOLD | FITTED | MOE | ||
| Top1 | < 1 Å | 40.6% | 19.8% | 32.9% | 32.4% | 16.4% | 23.7% | 20.3% | 14.5% |
| < 2 Å | 70.5% | 61.4% | 61.8% | 58.9% | 55.1% | 53.1% | 47.8% | 37.2% | |
| Top10 | < 1 Å | 61.8% | 37.7% | 42.0% | 49.8% | 29.5% | 37.2% | 39.6% | 22.7% |
| < 2 Å | 93.2% | 92.8% | 88.0% | 74.4% | 75.4% | 65.2% | 69.6% | 50.7% | |
| Type | RMSD | Success rates | |||||||
|---|---|---|---|---|---|---|---|---|---|
| HCovDock | Cov_DOX | ICM-Pro | CovDock | Autodock | GOLD | FITTED | MOE | ||
| Top1 | < 1 Å | 40.6% | 19.8% | 32.9% | 32.4% | 16.4% | 23.7% | 20.3% | 14.5% |
| < 2 Å | 70.5% | 61.4% | 61.8% | 58.9% | 55.1% | 53.1% | 47.8% | 37.2% | |
| Top10 | < 1 Å | 61.8% | 37.7% | 42.0% | 49.8% | 29.5% | 37.2% | 39.6% | 22.7% |
| < 2 Å | 93.2% | 92.8% | 88.0% | 74.4% | 75.4% | 65.2% | 69.6% | 50.7% | |
In addition to the top binding pose, we have also considered the top 10 ligand poses during the evaluations. The reason is 2-fold. On one hand, users may want to examine more than one binding pose of ligand during their experiments. On the other hand, users may also need more than one poses of ligand for further refinement and ranking with a more advanced scoring function during their computations. In such cases, a near-native ligand pose within a certain number of top predictions is necessary for further examinations. Table 1 lists the success rates of HCovDock and the other seven docking programs when the top 10 ligand poses are considered. It can be seen from the table that HCovDock also achieves the overall best performance among the eight docking methods. When the criterion of RMSD < 1.0 Å is used, HCovDock obtains the highest success rate of 61.8%, compared with 37.7% for Cov_DOX, 42.0% for ICM-Pro, 49.8% for CovDock, 29.5% for AutoDock, 37.2% for GOLD, 39.6% for FITTED and 22.7% for MOE. When the criterion of RMSD < 2.0 Å is adopted, HCovDock also achieves the best performance. These results demonstrate the robustness and accuracy of our HCovDock approach.
Impact factors on the docking performance
As shown in Figure 2, HCovDock achieves a significantly better performance than the other docking methods in reproducing experimental structures on the benchmark containing 207 complexes, and even outperforms some excellent docking algorithms. Furthermore, we investigate the influence of different factors on the docking performance, which includes the scoring function, the flexibility of the ligand and the type of warhead.
Scoring function
The most intrinsically difference between traditional docking and covalent docking lies in the covalent bond interactions between the ligand and its protein targets. Due to the covalent interaction, traditional scoring functions for non-covalent interactions will be no longer suitable for covalent docking. Therefore, covalent bond-based interactions should be considered to correct the scoring function, which is critical for the accuracy of covalent docking. As such, it is necessary to assess the impact of the covalent-induced scoring correction on the docking performance of HCovDock. Figure 3 shows the success rates of HCovDock as a function of the number of top predictions with/without considering the covalent bond-based scoring correction under the criteria of RMSD < 1.0 Å and < 2.0 Å, respectively. It can be seen from the figure that the docking success rate is greatly improved when HCovDock includes the covalent bond-based scoring correction. Specifically, HCovDock obtains the success rates of 40.6% and 70.5% for the top prediction under the criterion of RMSD < 1.0 Å and < 2.0 Å when including covalent bond-based scoring correction, which are much higher than 9.2% and 28.5% for the case without considering the scoring correction. This improvement can be understood as follows. The covalent bond-biased term set a threshold for VDW interactions, which reduces the penalty on the binding energy for the severe steric clashes between the receptor and the ligand, and allows the warhead segment to get closer to the receptor. That means, the covalent bond-biased term lowers the energy barrier for different conformations, and enhances the scoring accuracy of the warhead segment during sampling.
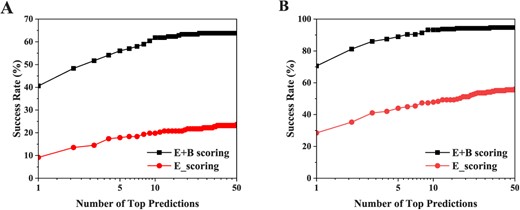
The influence of the covalent bond-induces scoring part on the docking performance under the RMSD criteria of < 1.0 Å(A) and < 2.0 Å(B). ‘E+B_scoring’ stands for the HCovDock with considering the covalent bond-based scoring energy correction. ‘E_scoring’ stands for the HCovDock without the covalent bond-based energy correction.
Ligand flexibility
In addition, we also investigate the effect of ligand flexibility on docking performance. According to the number of rotatable bonds, we defined three levels of ligands flexibility: ‘weak flexibility’ (|$\leqslant $| 10), ‘median flexibility’ (11–16) and ‘strong flexibility’(|$\geqslant $| 17). Figure 4 shows the success rates of HCovDock and the other seven covalent docking programs at different levels of ligand flexibility under the RMSD cutoff of 2.0 Å. As shown in Figure 4, all docking methods tend to yield lower success rates with the increase of the number of rotatable bonds when the top pose is considered (Figure 4A). This can be understood because it is more challenging to sample the conformations of a ligand with more rotatable bonds. Nevertheless, our HCovDock achieves an excellent docking accuracy and gives a high success rate of near or above 90.0% for different levels of flexible ligands when the top 10 predictions are considered (Figure 4B). That means that the ligand flexibility would not be a major limiting factor for the docking performance of HCovDock when the top 10 binding modes are considered. This is important for those users who want to examine more than one binding modes of ligand. Another feature in Figure 4 is that the docking performance of HCovDock is less ligand flexibility-dependent than that of other docking methods, suggesting the robustness of HCovDock.
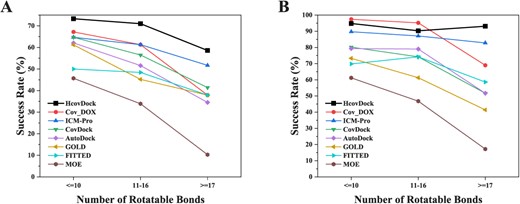
Impact of ligand flexibility on the covalent docking prediction in terms of the success rate versus the number of rotatable bonds of a ligand. The success rates of ‘weak flexibility’ (|$\leqslant $|10 rotatable bonds), ‘ median flexibility’ (11–16 rotatable bonds) and ‘strong flexibility’ (|$\geqslant $|17 rotatable bonds) for the top 1 (A) and top 10 (B) predictions are based on the RMSD criterion of <2.0 Å.
Warhead type
Furthermore, we explore the influence of different covalent bond formation modes on the docking performance. Figure 5A shows the docking success rates of HCovDock and the other seven docking algorithms for different warheads. It can be seen from the figure that HCovDock performs comparably well or better than the other docking methods for most warheads excepted the type of ‘Disulfide formation.’ Specifically, HCovDock obtains the success rates of 58.3%, 66.7%, 83.0%, 73.4%, 52.9%, 50.0% and 60.0% for Addition to Aldehyde, Addition to Ketone, Addition to Nitrile, Michael Addition, Nucleophilic Substitution, Ring Opening and Disulfide Formation prediction when the RMSD cutoff is 2Å, respectively, compared with 66.7%, 58.3%, 63.8%, 71.3%, 58.8%, 37.5% and 80.0% for the best performance of the other seven methods. Similar trends can be observed in the docking performances for top 10 predictions (Figure 5B). Relatively, HCovDock shows a comparatively worse performance in Aldehyde, Nucleophilic substitution and disulfide formation. Examinations for the corresponding complexes reveals that the worse performance in Aldehyde and Nucleophilic substitution is due to the caspase-3 cases that have a relatively open binding pocket as shown in target 2XYP (Figure 7a), which will pose a challenge for the scoring function in distinguishing the correct binding mode from a large number of sampled conformations. Further improvement may be needed to improve the docking accuracy for such types of covalent ligands. For disulfide formation, there are only five complexes in the test set. The small number of cases in the category of Disulfide Formation (only five complexes) would be not statistically enough to reflect the docking performance in this category.
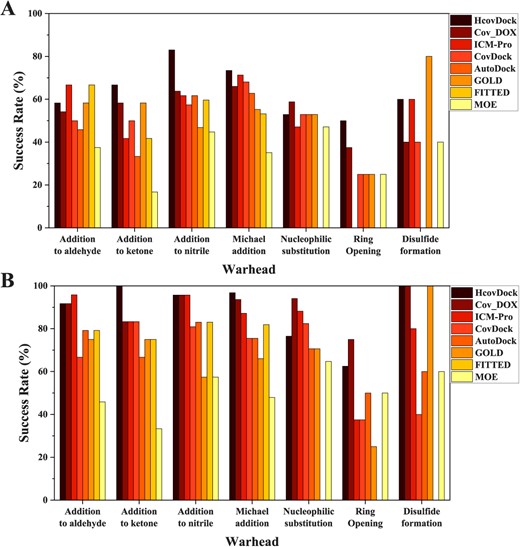
Comparison of the average success rates by HCovDock and the other seven docking methods for top 1 (A) and top 10 (B) predictions on different categories of warheads when the RMSD threshold is 2.0 Å.
Examples of predicted binding modes
Figure 6 shows two examples of the top ligand binding modes that are successfully predicted by HCovDock. Although the two complexes have a high flexible ligand with more than 20 rotatable bonds, HCovDock still achieves a docking accuracy of RMSD |$=1.51$| Å for the ligand of target 2CNO with 27 rotatable bonds (Figure 6A), and a docking accuracy of RMSD |$=1.41$| Å for the ligand of target 2AMD with 24 rotatable bonds (Figure 6C). Figure 6B and D shows the binding energy scores versus the RMSDs of the docked ligand poses for targets 2CNO and 2AMD, respectively. It can be seen from the two figures that the relationship between the energy scores and the RMSDs exhibits a good funnel shape, and the ligand poses with a lower RMSD tend to have a lower energy score. This suggests that the scoring function of HCovDock is robust to guide the docking process. The wide range of the RMSDs in Figures 6B and D also indicate that the binding conformations of ligand predicted by HCovDock are also diverse rather than constrained in a local minimum even, suggesting the good sampling diversity of HCovDock.
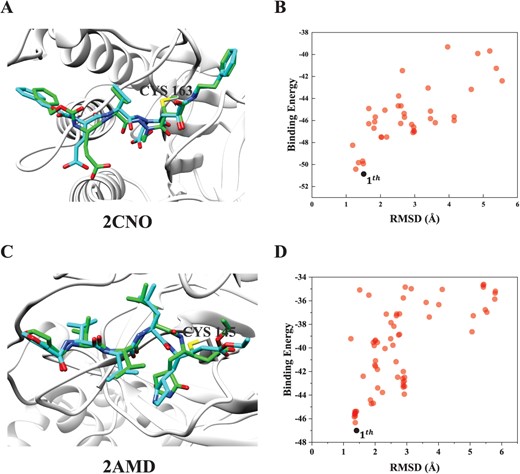
Comparison between the crystal structure (cyan) and HCovDock predicted pose (green) for two successfully docked complexes, where the protein is represented by molecular ribbons and the ligands are in stick mode. (A) Target 2CNO with a ligand containing 27 rotatable bonds, where the RMSD of the top pose is 1.51 Å. (B) The relationship between the binding scores and the RMSDs for top 100 binding conformations of 2CNO. (C) Target 2AMD with a ligand containing 23 rotatable bonds, where the RMSD of the top pose RMSD is 1.41 Å. (D) The relationship between the binding scores and the RMSDs for top 100 binding conformations of 2AMD.
Despite the good performance, HCovDock fails to predict correct poses on some cases. Figure 7A and B shows two failed docking examples (PDB IDs: 2XYP and 1THE), in which no near-native binding pose is predicted within the top 100 predictions. It can be seen from Figure 7A that the native ligand binds to the protein mainly through the hydrophobic interaction of two aromatic rings with the hydrophobic surface on the protein. However, for the predicted binding pose, one aromatic ring of the ligand is inserted into a well-defined pocket of the binding site for more atomic contacts between the ligand and the protein, even though the pocket is very hydrophilic. Such failure indicates that the hydrophobic interaction may be under-estimated in the scoring function of HCovDock. Similar phenomenon can be observed on Target 1THE. As shown in Figure 7B, the hydrophobic rings of the predicted pose are close to the hydrophilic surface of the protein. Such binding mode is in contrast to the native structure where the hydrophobic pars of the ligand form excellent hydrophobic interactions with the hydrophobic surface of the protein. These results suggest that the performance of HCovDock may be further improved by considering more hydrophobic interaction energy in the scoring function.
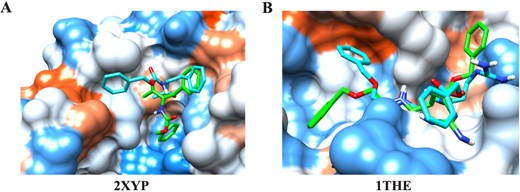
Comparison between the crystal structure (cyan) and the HCovDock predicted pose (green) for two failed docking cases. The protein is represented by a molecular surface and colored from red for the hydrophobic region to blue for the hydrophilic region. The ligands are in stick mode. (A) Target 2XYP. (B) Target 1THE.
Performance on an additional set of 81 complexes
To evaluate the general applicability of HCovDock, we tested the performance of HCovDock on an independent data set containing 81 complexes. The independent set is constructed from the covalent protein–ligand complexes collected by Xu et al. [70]. Specifically, the complex structures that satisfy the following conditions are preserved: 1) The resolution of complexes are better than 2.5 Å; 2) There are no missing residues on the protein structure within 6 Å neared the native ligand; 3) The complexes do not belong to the first test set of 207 cases. Thus, a total of 81 complex structures are obtained, which cover the above-mentioned seven types of reaction mechanisms between ligand and protein.
Table 2 shows the success rates of HCovDock in binding mode predictions under the criteria of RMSD < 1.0 Å and <2.0 Å on the independent benchmark of 81 covalent protein–ligand complexes when the ‘Top Pose’ and the ‘Best Pose’ are considered, respectively [34]. The detailed results for each complexes are listed in Supplementary Table S2. For comparison, Table 2 also lists the corresponding results of five other docking programs. It can be seen from the table that HCovDock obtains the success rates of 42.0% and 60.5% under the criterion of RMSD < 1.0 Å for the top pose and best pose, respectively, which are comparable to 40.6% and 61.8% on the first benchmark of 207 complexes. Comparing Tables 1 and 2 also reveals that the success rates on the independent set of 81 complexes tend to be lower than those on the first benchmark of 207 complexes under the criteria of RMSD < 2.0 Å for all docking approaches. Nevertheless, HCovDock still performed overall the best on the independent set of 81 complexes.
Comparison of the success rates of HCovDock and five other docking methods on an independent set of 81 complexes under two different RMSD criteria when the ‘Top Pose’ and the ‘Best Pose’ are considered, respectively, where the results of the methods other than HCoveDock are taken from the literature [34]. The number in bold font indicates the highest success rate for the category.
| Type | RMSD | Success rates | |||||
|---|---|---|---|---|---|---|---|
| HCovDock | Cov_DOX | ICM-Pro | CovDock | GOLD | MOE | ||
| Top Posea | < 1 Å | 42.0% | 22.2% | 27.2% | 29.6% | 19.8% | 2.5% |
| < 2 Å | 65.4% | 61.7% | 49.6% | 64.2% | 54.3% | 54.3% | |
| Best Poseb | < 1 Å | 60.5% | 42.0% | 27.2% | 35.8% | 30.9% | 8.6% |
| < 2 Å | 85.2% | 88.9% | 64.2% | 75.3% | 64.2% | 77.8% | |
| Type | RMSD | Success rates | |||||
|---|---|---|---|---|---|---|---|
| HCovDock | Cov_DOX | ICM-Pro | CovDock | GOLD | MOE | ||
| Top Posea | < 1 Å | 42.0% | 22.2% | 27.2% | 29.6% | 19.8% | 2.5% |
| < 2 Å | 65.4% | 61.7% | 49.6% | 64.2% | 54.3% | 54.3% | |
| Best Poseb | < 1 Å | 60.5% | 42.0% | 27.2% | 35.8% | 30.9% | 8.6% |
| < 2 Å | 85.2% | 88.9% | 64.2% | 75.3% | 64.2% | 77.8% | |
aThe ‘Top Pose’ refers to the top-ranked binding pose according to its score or its calculated energy. bThe ‘Best Pose’ refers to the most crystal-structure like pose in the conformation space regardless of its score or energy. The success rate of the ‘Best Pose’ is based on the top 10 poses for HCovDock and Cov_DOX, and all the sampled poses for the other methods.
Comparison of the success rates of HCovDock and five other docking methods on an independent set of 81 complexes under two different RMSD criteria when the ‘Top Pose’ and the ‘Best Pose’ are considered, respectively, where the results of the methods other than HCoveDock are taken from the literature [34]. The number in bold font indicates the highest success rate for the category.
| Type | RMSD | Success rates | |||||
|---|---|---|---|---|---|---|---|
| HCovDock | Cov_DOX | ICM-Pro | CovDock | GOLD | MOE | ||
| Top Posea | < 1 Å | 42.0% | 22.2% | 27.2% | 29.6% | 19.8% | 2.5% |
| < 2 Å | 65.4% | 61.7% | 49.6% | 64.2% | 54.3% | 54.3% | |
| Best Poseb | < 1 Å | 60.5% | 42.0% | 27.2% | 35.8% | 30.9% | 8.6% |
| < 2 Å | 85.2% | 88.9% | 64.2% | 75.3% | 64.2% | 77.8% | |
| Type | RMSD | Success rates | |||||
|---|---|---|---|---|---|---|---|
| HCovDock | Cov_DOX | ICM-Pro | CovDock | GOLD | MOE | ||
| Top Posea | < 1 Å | 42.0% | 22.2% | 27.2% | 29.6% | 19.8% | 2.5% |
| < 2 Å | 65.4% | 61.7% | 49.6% | 64.2% | 54.3% | 54.3% | |
| Best Poseb | < 1 Å | 60.5% | 42.0% | 27.2% | 35.8% | 30.9% | 8.6% |
| < 2 Å | 85.2% | 88.9% | 64.2% | 75.3% | 64.2% | 77.8% | |
aThe ‘Top Pose’ refers to the top-ranked binding pose according to its score or its calculated energy. bThe ‘Best Pose’ refers to the most crystal-structure like pose in the conformation space regardless of its score or energy. The success rate of the ‘Best Pose’ is based on the top 10 poses for HCovDock and Cov_DOX, and all the sampled poses for the other methods.
Comparison of the AUC in virtual screening by HCovDock and the three scoring schemes of CovDock on 10 protein receptors, where the results of CovDock are taken from the literature [32].
| PDB ID | Target | AUC | |||
|---|---|---|---|---|---|
| HCovDock | CovDock-prea | CovDock-postb | CovDock-averagec | ||
| 2hwo | cSrc | 0.736 | 0.677 | 0.626 | 0.780 |
| 2hwp | cSrc | 0.756 | 0.804 | 0.873 | 0.858 |
| 2qlq | cSrc | 0.878 | 0.802 | 0.835 | 0.825 |
| 2qq7 | cSrc | 0.753 | 0.732 | 0.824 | 0.836 |
| 3lok | cSrc | 0.785 | 0.956 | 0.816 | 0.864 |
| 2j5e | EGFR | 0.888 | 0.862 | 0.955 | 0.950 |
| 2j5f | EGFR | 0.845 | 0.764 | 0.842 | 0.829 |
| 2jiv | EGFR | 0.701 | 0.942 | 0.906 | 0.941 |
| 3ika | EGFR | 0.875 | 0.614 | 0.735 | 0.701 |
| 3v6s | JNK | 0.758 | 0.448 | 0.395 | 0.428 |
| Average | 0.798 | 0.760 | 0.781 | 0.801 | |
| PDB ID | Target | AUC | |||
|---|---|---|---|---|---|
| HCovDock | CovDock-prea | CovDock-postb | CovDock-averagec | ||
| 2hwo | cSrc | 0.736 | 0.677 | 0.626 | 0.780 |
| 2hwp | cSrc | 0.756 | 0.804 | 0.873 | 0.858 |
| 2qlq | cSrc | 0.878 | 0.802 | 0.835 | 0.825 |
| 2qq7 | cSrc | 0.753 | 0.732 | 0.824 | 0.836 |
| 3lok | cSrc | 0.785 | 0.956 | 0.816 | 0.864 |
| 2j5e | EGFR | 0.888 | 0.862 | 0.955 | 0.950 |
| 2j5f | EGFR | 0.845 | 0.764 | 0.842 | 0.829 |
| 2jiv | EGFR | 0.701 | 0.942 | 0.906 | 0.941 |
| 3ika | EGFR | 0.875 | 0.614 | 0.735 | 0.701 |
| 3v6s | JNK | 0.758 | 0.448 | 0.395 | 0.428 |
| Average | 0.798 | 0.760 | 0.781 | 0.801 | |
aCovDock-pre refers to the Glide score for the pre-reactive binding mode. bCovDock-post refers to the Glide score for the post-reactive binding mode. cCovDock-average refers to the average Glide scores for the pre- and post-reactive binding modes.
Comparison of the AUC in virtual screening by HCovDock and the three scoring schemes of CovDock on 10 protein receptors, where the results of CovDock are taken from the literature [32].
| PDB ID | Target | AUC | |||
|---|---|---|---|---|---|
| HCovDock | CovDock-prea | CovDock-postb | CovDock-averagec | ||
| 2hwo | cSrc | 0.736 | 0.677 | 0.626 | 0.780 |
| 2hwp | cSrc | 0.756 | 0.804 | 0.873 | 0.858 |
| 2qlq | cSrc | 0.878 | 0.802 | 0.835 | 0.825 |
| 2qq7 | cSrc | 0.753 | 0.732 | 0.824 | 0.836 |
| 3lok | cSrc | 0.785 | 0.956 | 0.816 | 0.864 |
| 2j5e | EGFR | 0.888 | 0.862 | 0.955 | 0.950 |
| 2j5f | EGFR | 0.845 | 0.764 | 0.842 | 0.829 |
| 2jiv | EGFR | 0.701 | 0.942 | 0.906 | 0.941 |
| 3ika | EGFR | 0.875 | 0.614 | 0.735 | 0.701 |
| 3v6s | JNK | 0.758 | 0.448 | 0.395 | 0.428 |
| Average | 0.798 | 0.760 | 0.781 | 0.801 | |
| PDB ID | Target | AUC | |||
|---|---|---|---|---|---|
| HCovDock | CovDock-prea | CovDock-postb | CovDock-averagec | ||
| 2hwo | cSrc | 0.736 | 0.677 | 0.626 | 0.780 |
| 2hwp | cSrc | 0.756 | 0.804 | 0.873 | 0.858 |
| 2qlq | cSrc | 0.878 | 0.802 | 0.835 | 0.825 |
| 2qq7 | cSrc | 0.753 | 0.732 | 0.824 | 0.836 |
| 3lok | cSrc | 0.785 | 0.956 | 0.816 | 0.864 |
| 2j5e | EGFR | 0.888 | 0.862 | 0.955 | 0.950 |
| 2j5f | EGFR | 0.845 | 0.764 | 0.842 | 0.829 |
| 2jiv | EGFR | 0.701 | 0.942 | 0.906 | 0.941 |
| 3ika | EGFR | 0.875 | 0.614 | 0.735 | 0.701 |
| 3v6s | JNK | 0.758 | 0.448 | 0.395 | 0.428 |
| Average | 0.798 | 0.760 | 0.781 | 0.801 | |
aCovDock-pre refers to the Glide score for the pre-reactive binding mode. bCovDock-post refers to the Glide score for the post-reactive binding mode. cCovDock-average refers to the average Glide scores for the pre- and post-reactive binding modes.
Virtual screening
We further evaluate the power of the virtual screening of HCovDock on a test set of 10 receptors. This test set is challenging due to its high similarity between true binders and decoy ligands [32]. Table 3 shows the areas under the curves (AUC) of HCovDock in virtual screening on the 10 receptors. For comparison, the table also lists the corresponding results of the three scoring schemes by CovDock – a covalent docking protocol adapted from Glide in the Schrödinger docking suite [33]. It can be seen from Table 3 that HCovDock performs better than the pre- and post-scoring schemes of CovDock, and comparably well with its average scoring scheme. Specifically, HCovDock obtains an average AUC of 0.798 on the 10 protein tarters, compared with 0.760 for CovDock-pre, 0.781 for CovDock-post and 0.801 for CovDock-average. As a virtual screening tool, its robustness is also critical and will affect the generalization ability of virtual screening on different receptors. Table 3 shows that the AUC of HCovDock ranges from 0.701 to 0.888, compared with the range from 0.395 to 0.956 for CovDock. It is also worth mentioning that the energy and conformation predicted by HCovDock are in a one-to-one correspondence, i.e. the optimization of the conformation can be directly guided by the binding energy score. However, the CovDock-average scoring scheme adopts a combined strategy to evaluate their docking results and may not rank the covalent inhibitors with much different intrinsic reactivities well [32].
Computational efficiency
The computational efficiency of a protein–ligand docking program is also crucial, especially for the case of virtual screening where a large number of ligands need to be docked and evaluated. Figure 8 shows the average running time of HCovDock as a function of the number of rotatable bonds for the covalent docking on the benchmark of 207 complexes. Here, all the docking calculations are conducted on a single CPU core on a Linux system with the Intel(R) Xeon E5-2690 v3 2.60 GHz CPU. It can be seen from the figure that HCovDock tends to consume more time for the molecules with more rotatable bonds, and finishes a covalent docking job within as short as 0.27 s for target 4THI with a ligand containing one rotatable bond. The longest running time is 17.7 min for target 3ZVG with a ligand containing 23 rotatable bonds. This can be understood because the HCovDock needs more time to explore the sampling space for more flexible ligands. The running time has a high correlation with the number of rotatable bonds of the ligand (|$R=$| 0.825), suggesting that the number of rotatable bonds of the ligand can be a good estimate of the running time for covalent docking with HCovDock. On average, HCovDock consumes about 5.0 min for a covalent docking job on a single CPU core. As a comparison, two other programs, Cov_DOX(PM7) and AutoDock roughly takes 0.5–1 h and 1 h for completing a docking job, respectively. These results suggest the high computational efficiency of HCovDock.

The running time of HCovDock versus the number of rotatable bonds of the ligand on the test set of 207 complexes. The red line is the linear fitting of the relationship.
Conclusion
We have developed a covalent docking algorithm, named HCovDock, to predict the binding conformations of covalent protein–ligand complexes. Based on the search method of incremental construction, HCovDock employs a pruning strategy and a covalent bond-based scoring correction to balance docking accuracy and computational efficiency. With the RMSD threshold of 1.0 Å, the success rates for top 1 and top 10 predictions given by our HCovDock are 40.6 % and 61.8 % on a benchmark of 207 covalent protein–ligand complexes, respectively. When the RMSD cutoff is set to 2.0 Å, the success rates for top 1 and top 10 predictions obtained by HCovDock can reach 70.5 % and 93.2 %. HCovDock is also compared with seven state-of-the-art other covalent docking programs, and exhibits a significantly better performance compared with the other docking programs. For validating the virtual screening ability, HCovDock is tested on the 10 receptors of three proteins and achieved an average AUC of 0.798. In addition to a good docking accuracy, HCovDock is also efficient in terms of computational time, with an average running time of 5.0 min. Given the excellent docking performance and high computational efficiency, HCovDock is expected to serve as a valuable docking tool in covalent drug discovery.
Despite its simple scoring function, HCovDock achieves significant advantages over existing methods, which can be attributed to several aspects. One major difference between HCovDock and the other methods is the introduction of a layer-dependent covalent bond-based energy correction, which yields a good balance between covalent and non-covalent interactions and thus allow the ligand poses to be ranked more reasonably. In addition, unlike other methods which use stochastic algorithms like MD/MC simulations or Genetic approaches to consider the ligand flexibility during the docking calculations, HCovDock adopts an incremental construction method to build the ligand binding conformation under the restriction of the binding pocket, which will allow more efficient ligand binding conformational sampling and enable the ligand to better fit the binding site. Moreover, the flexibility of some challenging fragments like rings in the ligand can be further considered by docking the multiple ligand conformations generated by the Conformator and OpenBabel programs.
Covalent inhibitors have received extensive attentions in drug discovery.
We have developed an efficient covalent ligand docking algorithm HCovDock.
HCovDock integrates incremental construction and covalent bond-based energy.
HCovDock much outperformed existing approaches in binding mode predictions.
HCovDock is fast and can finish docking a ligand within minutes.
Data and Software Availability
The HCovDock server can be accessed at http://huanglab.phys.hust.edu.cn/hcovdock/. The data in this study are available from the corresponding author upon request.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant Nos 62072199 and 32161133002) and the startup grant of Huazhong University of Science and Technology.
Author Biographies
Qilong Wu is a PhD student in the School of Physics at Huazhong University of Science and Technology. His research interests include molecular docking and scoring function for protein–ligand interactions.
Sheng-You Huang is a full professor in the School of Physics at the Huazhong University of Science and Technology. His research interests are molecular docking and scoring functions for protein–ligand interactions, protein–protein interactions and protein–peptide interactions. More information about his research group can be found at http://huanglab.phys.hust.edu.cn/.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}