




Abstract
Internal validation is the most popular evaluation strategy used for drug–target predictive models. The simple random shuffling in the cross-validation, however, is not always ideal to handle large, diverse and copious datasets as it could potentially introduce bias. Hence, these predictive models cannot be comprehensively evaluated to provide insight into their general performance on a variety of use-cases (e.g. permutations of different levels of connectiveness and categories in drug and target space, as well as validations based on different data sources). In this work, we introduce a benchmark, BETA, that aims to address this gap by (i) providing an extensive multipartite network consisting of 0.97 million biomedical concepts and 8.5 million associations, in addition to 62 million drug–drug and protein–protein similarities and (ii) presenting evaluation strategies that reflect seven cases (i.e. general, screening with different connectivity, target and drug screening based on categories, searching for specific drugs and targets and drug repurposing for specific diseases), a total of seven Tests (consisting of 344 Tasks in total) across multiple sampling and validation strategies. Six state-of-the-art methods covering two broad input data types (chemical structure- and gene sequence-based and network-based) were tested across all the developed Tasks. The best-worst performing cases have been analyzed to demonstrate the ability of the proposed benchmark to identify limitations of the tested methods for running over the benchmark tasks. The results highlight BETA as a benchmark in the selection of computational strategies for drug repurposing and target discovery.
Introduction
Critical to the drug discovery process is the ability to define, identify, screen and understand potential candidate pairs among small molecules (i.e. drugs) and proteins (i.e. targets) [1–3]. Despite advancements in the use of biological assays to experimentally validate drug–target interactions (DTIs), these early steps of drug development remain expensive to accomplish [3, 4]. The use of current experimental screening (in vitro) methods to cover all the possible combinations of DTIs is infeasible, and the tendency to only focus on particular families of ‘druggable’ proteins or ‘preferable’ drugs greatly limits the systematic screening of the potentially larger number of compounds, small molecules and proteins available [5, 6]. The adoption of computational (in silico) methods has therefore been suggested to provide a more efficient means for prescreening [7–14].
Computational methods historically began with early attempts of docking simulations and ligand matching [2, 3, 15, 16] and have recently progressed to machine learning-based solutions [14, 17–20]. Although it is desirable to validate the discoveries with biological assays, known as external validation, it is infeasible for most computational labs. Therefore, internal validation, such as cross-validation, is the most popular validation strategy for the existing methods, where some of the drug–target associations remain for testing during the training process. The datasets in internal validations are either small-scale datasets developed from very early attempts [21–25] or tailored sets generated from diverse biomedical databases that contain drug–target associations [14]. For example, among the 87 investigated computational papers published in a recent survey [14], 79 (91%) and 66 (77%) papers conducted the experiments based on the biomedical databases Drugbank [26] and Kyoto Encyclopedia of Genes and Genomes (KEGG) [27], and 54 (62%) used small-scale drug–target associations based on a protein category in the target space developed for cross-validation in 2008 [28]. Bias may still exist in these experiments with simple random suffering as the patterns of connectiveness and categories in the drug and target space with a large number of associations will be favored. A gold standard that provides large datasets, as well as sophisticated validation methods with a minimized risk of bias (e.g. permutations of different levels of connectiveness and categories in the drug and target space, as well as validations based on different data sources), does not exist. The complexity in selecting suitable computational solutions during the drug development phase is an ongoing challenge [21, 23, 29, 30], and without such a standard benchmark to evaluate predictive models in an equitable and comprehensive manner, the adoptability of developed computational methods is hindered.
In this work, we fill this gap by providing a large-scale benchmark that enables a comprehensive evaluation of drug–target predictive models to facilitate a selection of computational strategies for drug and target prescreening. This benchmark provides an extensive multipartite network consisting of 0.97 million biomedical concepts including 59 000 drugs and 95 000 targets, and 8.5 million associations including 817 thousand drug–target associations, as well as 62 million drug–drug and protein–protein similarities based on drug chemical structures and gene sequences that can be used to comprehensively evaluate the prescreening strategies that reflect seven use-cases (i.e. general, screening with different connectivity, target and drug screening based on categories, searching for specific drugs and targets and drug repurposing for specific diseases), a total of seven Tests (consisting of 344 Tasks in total) that cover two types of training/testing sampling strategies based on drug–target space as well as six types of validation strategies. To demonstrate the use of our benchmark, six state-of-the-art predictive models have been selected and categorized based on the input types (i.e. structure- and sequence-based and network-based methods) and evaluated as use-cases. The best-worst performing diseases (e.g. spinal muscular atrophy versus obesity for a versioning-based Job and human immunodeficiency virus (HIV) versus myocardial infarction for a trial-based Job) have been analyzed. The results highlighted BETA as a benchmark in the selection of drug–target prediction methods for drug repurposing and target discovery applications when a pair of drugs and targets are given as the input.
Methods
Our proposed benchmark consists of two major components: (i) datasets and (ii) evaluation Tasks.
With respect to datasets, a multipartite network was constructed based on an integration of 11 existing biomedical repositories (Diseasome [31], Drugbank [26], Gene Ontology Annotation (GOA) [32], Interaction Reference Index (iRefindex) [33], KEGG [27], Linked Structured Product Label (Linkedspl) [34], Online Mendelian Inheritance in Man (OMIM) [35], Pharmacogenomics Knowledge Base (Pharmgkb) [36], Side Effect Resource (SIDER) [37] and STRING [38]), which incorporated 971 874 entities and 8 530 037 associations in total. We defined a common ‘drug–target–disease’ node space that consisted of the entities from Drugbank (6250 drugs and 2838 targets) and OMIM (52 187 diseases) (see Table 1(a) for details). This graph also incorporated 46 million drug–drug and 16 million protein–protein similarities computed based on the chemical structures and gene sequence obtained from Drugbank.
Statistics of the benchmark dataset and evaluation Tasks
| (a) Network | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Repositories | Node (biomedical entities) | Edge (biomedical associations) | Drug mapped | Target mapped | Disease mapped | |||||||||
| All node | Drug | Target | Disease | All edge | Drug-associated | Target-associated | Disease-associated | Local | Drugbank | Local | Drugbank | Local | OMIM | |
| Diseasome | 9518 | 1362 | 3919 | 4213 | 25 091 | 8202 | 9744 | 268 918 | 1309 | 1309 | 508 | 515 | 445 | 557 |
| DrugBank | 24 655 | 6823 | 4037 | 0 | 56 245 | 56 245 | 14 744 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| GOA | 48 624 | 0 | 43 830 | 0 | 163 498 | 0 | 163 715 | 0 | 0 | 0 | 2178 | 2180 | 0 | 0 |
| Irefindex | 230 136 | 0 | 14 860 | 0 | 59 809 | 0 | 975 287 | 0 | 0 | 0 | 2555 | 2562 | 0 | 0 |
| KEGG | 13 776 | 3848 | 7777 | 1284 | 975 287 | 58 476 | 45 465 | 35 725 | 2380 | 1676 | 2165 | 2159 | 1036 | 2751 |
| Linkedspl | 61 869 | 44 196 | 30 | 0 | 163 639 | 163 639 | 3613 | 0 | 37 807 | 1134 | 2131 | 2140 | 0 | 0 |
| OMIM | 104 540 | 0 | 0 | 31 468 | 137 682 | 0 | 0 | 137 682 | 0 | 0 | 2118 | 2120 | 0 | 0 |
| Pharmgkb | 6543 | 868 | 1442 | 486 | 33 508 | 23 322 | 17 610 | 25 460 | 1643 | 1638 | 2131 | 2140 | 1376 | 2208 |
| Pharmgkb-offside | 450 230 | 1332 | 0 | 10 097 | 877 604 | 438 802 | 0 | 438 802 | 7149 | 6138 | 0 | 0 | 50 905 | 52 159 |
| Sider | 2598 | 893 | 0 | 1705 | 68 424 | 68 424 | 0 | 68 424 | 858 | 868 | 0 | 0 | 814 | 1428 |
| STRING | 19 385 | 0 | 19 385 | 0 | 5 969 250 | 0 | 5 969 250 | 0 | 0 | 0 | 2131 | 2133 | 0 | 0 |
| Total | 971 874 | 59 322 | 95 280 | 49 253 | 8 530 037 | 817 110 | 7 199 428 | 975 011 | 51 146 | 6250 | 15 917 | 2838 | 54 576 | 52 187 |
| (b) Evaluation tasks | ||||||||||||||
| Test | Purpose | Validation | Drug–Target Space | # Task | # Avg train pairs | # Avg drugs for training | # Avg targets for training | # Avg test pairs | # Avg test positive | # Avg test negative | # Avg drugs for testing | # Avg targets for testing | ||
| 0 | General | k-fold cross internal validation (k = 10) | All | 10 | 11 016 | 5337 | 3087 | 1483 | 676 | 807 | 1190 | 1039 | ||
| 1 | Screening with connectivity spaces (e.g. isolated drugs or targets) | k-fold cross internal validation (k = 10) | Connectivity space | 90 | 11 769 | 5654 | 3221 | 929 | 471 | 458 | 637 | 551 | ||
| 2 | Target screening when drugs are within/beyond category | k-fold cross internal validation (k = 3) + Drug categories | Category space (drugbank category) | 36 | 12 164 | 5731 | 3252 | 152 | 76 | 76 | 32 | 73 | ||
| Category space (linkpl class) | 36 | 12 108 | 5732 | 3201 | 263 | 132 | 132 | 32 | 192 | |||||
| 3 | Drug screening when targets are within/beyond category | k-fold cross internal validation (k = 3) + Protein categories | Category space (family) | 36 | 12 045 | 5721 | 3252 | 389 | 195 | 195 | 109 | 120 | ||
| Category space (protein class) | 36 | 11 965 | 5685 | 3247 | 549 | 275 | 275 | 165 | 194 | |||||
| 4 | Searching for a specific drug/target | Internal validation with m search (n = 10) | All targets (500) | 10 | 12 207 | 5735 | 3239 | 534 | 33 | 501 | 1 | 534 | ||
| Internal validation with m search (n = 10) | All drugs (500) | 10 | 12 196 | 5717 | 3253 | 545 | 44 | 501 | 545 | 1 | ||||
| 5 | Drug repurposing for a specific disease | Version-based external validation + Disease specific | Category space (10 disease) | 40 | 12 240 | 5735 | 3253 | 44 | 22 | 22 | 24 | 14 | ||
| 6 | Clinical trial-based external validation + Disease-specific | Category space (10 disease) | 40 | 12 240 | 5735 | 3253 | 25 | 13 | 13 | 10 | 11 | |||
| (a) Network | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Repositories | Node (biomedical entities) | Edge (biomedical associations) | Drug mapped | Target mapped | Disease mapped | |||||||||
| All node | Drug | Target | Disease | All edge | Drug-associated | Target-associated | Disease-associated | Local | Drugbank | Local | Drugbank | Local | OMIM | |
| Diseasome | 9518 | 1362 | 3919 | 4213 | 25 091 | 8202 | 9744 | 268 918 | 1309 | 1309 | 508 | 515 | 445 | 557 |
| DrugBank | 24 655 | 6823 | 4037 | 0 | 56 245 | 56 245 | 14 744 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| GOA | 48 624 | 0 | 43 830 | 0 | 163 498 | 0 | 163 715 | 0 | 0 | 0 | 2178 | 2180 | 0 | 0 |
| Irefindex | 230 136 | 0 | 14 860 | 0 | 59 809 | 0 | 975 287 | 0 | 0 | 0 | 2555 | 2562 | 0 | 0 |
| KEGG | 13 776 | 3848 | 7777 | 1284 | 975 287 | 58 476 | 45 465 | 35 725 | 2380 | 1676 | 2165 | 2159 | 1036 | 2751 |
| Linkedspl | 61 869 | 44 196 | 30 | 0 | 163 639 | 163 639 | 3613 | 0 | 37 807 | 1134 | 2131 | 2140 | 0 | 0 |
| OMIM | 104 540 | 0 | 0 | 31 468 | 137 682 | 0 | 0 | 137 682 | 0 | 0 | 2118 | 2120 | 0 | 0 |
| Pharmgkb | 6543 | 868 | 1442 | 486 | 33 508 | 23 322 | 17 610 | 25 460 | 1643 | 1638 | 2131 | 2140 | 1376 | 2208 |
| Pharmgkb-offside | 450 230 | 1332 | 0 | 10 097 | 877 604 | 438 802 | 0 | 438 802 | 7149 | 6138 | 0 | 0 | 50 905 | 52 159 |
| Sider | 2598 | 893 | 0 | 1705 | 68 424 | 68 424 | 0 | 68 424 | 858 | 868 | 0 | 0 | 814 | 1428 |
| STRING | 19 385 | 0 | 19 385 | 0 | 5 969 250 | 0 | 5 969 250 | 0 | 0 | 0 | 2131 | 2133 | 0 | 0 |
| Total | 971 874 | 59 322 | 95 280 | 49 253 | 8 530 037 | 817 110 | 7 199 428 | 975 011 | 51 146 | 6250 | 15 917 | 2838 | 54 576 | 52 187 |
| (b) Evaluation tasks | ||||||||||||||
| Test | Purpose | Validation | Drug–Target Space | # Task | # Avg train pairs | # Avg drugs for training | # Avg targets for training | # Avg test pairs | # Avg test positive | # Avg test negative | # Avg drugs for testing | # Avg targets for testing | ||
| 0 | General | k-fold cross internal validation (k = 10) | All | 10 | 11 016 | 5337 | 3087 | 1483 | 676 | 807 | 1190 | 1039 | ||
| 1 | Screening with connectivity spaces (e.g. isolated drugs or targets) | k-fold cross internal validation (k = 10) | Connectivity space | 90 | 11 769 | 5654 | 3221 | 929 | 471 | 458 | 637 | 551 | ||
| 2 | Target screening when drugs are within/beyond category | k-fold cross internal validation (k = 3) + Drug categories | Category space (drugbank category) | 36 | 12 164 | 5731 | 3252 | 152 | 76 | 76 | 32 | 73 | ||
| Category space (linkpl class) | 36 | 12 108 | 5732 | 3201 | 263 | 132 | 132 | 32 | 192 | |||||
| 3 | Drug screening when targets are within/beyond category | k-fold cross internal validation (k = 3) + Protein categories | Category space (family) | 36 | 12 045 | 5721 | 3252 | 389 | 195 | 195 | 109 | 120 | ||
| Category space (protein class) | 36 | 11 965 | 5685 | 3247 | 549 | 275 | 275 | 165 | 194 | |||||
| 4 | Searching for a specific drug/target | Internal validation with m search (n = 10) | All targets (500) | 10 | 12 207 | 5735 | 3239 | 534 | 33 | 501 | 1 | 534 | ||
| Internal validation with m search (n = 10) | All drugs (500) | 10 | 12 196 | 5717 | 3253 | 545 | 44 | 501 | 545 | 1 | ||||
| 5 | Drug repurposing for a specific disease | Version-based external validation + Disease specific | Category space (10 disease) | 40 | 12 240 | 5735 | 3253 | 44 | 22 | 22 | 24 | 14 | ||
| 6 | Clinical trial-based external validation + Disease-specific | Category space (10 disease) | 40 | 12 240 | 5735 | 3253 | 25 | 13 | 13 | 10 | 11 | |||
Statistics of the benchmark dataset and evaluation Tasks
| (a) Network | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Repositories | Node (biomedical entities) | Edge (biomedical associations) | Drug mapped | Target mapped | Disease mapped | |||||||||
| All node | Drug | Target | Disease | All edge | Drug-associated | Target-associated | Disease-associated | Local | Drugbank | Local | Drugbank | Local | OMIM | |
| Diseasome | 9518 | 1362 | 3919 | 4213 | 25 091 | 8202 | 9744 | 268 918 | 1309 | 1309 | 508 | 515 | 445 | 557 |
| DrugBank | 24 655 | 6823 | 4037 | 0 | 56 245 | 56 245 | 14 744 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| GOA | 48 624 | 0 | 43 830 | 0 | 163 498 | 0 | 163 715 | 0 | 0 | 0 | 2178 | 2180 | 0 | 0 |
| Irefindex | 230 136 | 0 | 14 860 | 0 | 59 809 | 0 | 975 287 | 0 | 0 | 0 | 2555 | 2562 | 0 | 0 |
| KEGG | 13 776 | 3848 | 7777 | 1284 | 975 287 | 58 476 | 45 465 | 35 725 | 2380 | 1676 | 2165 | 2159 | 1036 | 2751 |
| Linkedspl | 61 869 | 44 196 | 30 | 0 | 163 639 | 163 639 | 3613 | 0 | 37 807 | 1134 | 2131 | 2140 | 0 | 0 |
| OMIM | 104 540 | 0 | 0 | 31 468 | 137 682 | 0 | 0 | 137 682 | 0 | 0 | 2118 | 2120 | 0 | 0 |
| Pharmgkb | 6543 | 868 | 1442 | 486 | 33 508 | 23 322 | 17 610 | 25 460 | 1643 | 1638 | 2131 | 2140 | 1376 | 2208 |
| Pharmgkb-offside | 450 230 | 1332 | 0 | 10 097 | 877 604 | 438 802 | 0 | 438 802 | 7149 | 6138 | 0 | 0 | 50 905 | 52 159 |
| Sider | 2598 | 893 | 0 | 1705 | 68 424 | 68 424 | 0 | 68 424 | 858 | 868 | 0 | 0 | 814 | 1428 |
| STRING | 19 385 | 0 | 19 385 | 0 | 5 969 250 | 0 | 5 969 250 | 0 | 0 | 0 | 2131 | 2133 | 0 | 0 |
| Total | 971 874 | 59 322 | 95 280 | 49 253 | 8 530 037 | 817 110 | 7 199 428 | 975 011 | 51 146 | 6250 | 15 917 | 2838 | 54 576 | 52 187 |
| (b) Evaluation tasks | ||||||||||||||
| Test | Purpose | Validation | Drug–Target Space | # Task | # Avg train pairs | # Avg drugs for training | # Avg targets for training | # Avg test pairs | # Avg test positive | # Avg test negative | # Avg drugs for testing | # Avg targets for testing | ||
| 0 | General | k-fold cross internal validation (k = 10) | All | 10 | 11 016 | 5337 | 3087 | 1483 | 676 | 807 | 1190 | 1039 | ||
| 1 | Screening with connectivity spaces (e.g. isolated drugs or targets) | k-fold cross internal validation (k = 10) | Connectivity space | 90 | 11 769 | 5654 | 3221 | 929 | 471 | 458 | 637 | 551 | ||
| 2 | Target screening when drugs are within/beyond category | k-fold cross internal validation (k = 3) + Drug categories | Category space (drugbank category) | 36 | 12 164 | 5731 | 3252 | 152 | 76 | 76 | 32 | 73 | ||
| Category space (linkpl class) | 36 | 12 108 | 5732 | 3201 | 263 | 132 | 132 | 32 | 192 | |||||
| 3 | Drug screening when targets are within/beyond category | k-fold cross internal validation (k = 3) + Protein categories | Category space (family) | 36 | 12 045 | 5721 | 3252 | 389 | 195 | 195 | 109 | 120 | ||
| Category space (protein class) | 36 | 11 965 | 5685 | 3247 | 549 | 275 | 275 | 165 | 194 | |||||
| 4 | Searching for a specific drug/target | Internal validation with m search (n = 10) | All targets (500) | 10 | 12 207 | 5735 | 3239 | 534 | 33 | 501 | 1 | 534 | ||
| Internal validation with m search (n = 10) | All drugs (500) | 10 | 12 196 | 5717 | 3253 | 545 | 44 | 501 | 545 | 1 | ||||
| 5 | Drug repurposing for a specific disease | Version-based external validation + Disease specific | Category space (10 disease) | 40 | 12 240 | 5735 | 3253 | 44 | 22 | 22 | 24 | 14 | ||
| 6 | Clinical trial-based external validation + Disease-specific | Category space (10 disease) | 40 | 12 240 | 5735 | 3253 | 25 | 13 | 13 | 10 | 11 | |||
| (a) Network | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Repositories | Node (biomedical entities) | Edge (biomedical associations) | Drug mapped | Target mapped | Disease mapped | |||||||||
| All node | Drug | Target | Disease | All edge | Drug-associated | Target-associated | Disease-associated | Local | Drugbank | Local | Drugbank | Local | OMIM | |
| Diseasome | 9518 | 1362 | 3919 | 4213 | 25 091 | 8202 | 9744 | 268 918 | 1309 | 1309 | 508 | 515 | 445 | 557 |
| DrugBank | 24 655 | 6823 | 4037 | 0 | 56 245 | 56 245 | 14 744 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| GOA | 48 624 | 0 | 43 830 | 0 | 163 498 | 0 | 163 715 | 0 | 0 | 0 | 2178 | 2180 | 0 | 0 |
| Irefindex | 230 136 | 0 | 14 860 | 0 | 59 809 | 0 | 975 287 | 0 | 0 | 0 | 2555 | 2562 | 0 | 0 |
| KEGG | 13 776 | 3848 | 7777 | 1284 | 975 287 | 58 476 | 45 465 | 35 725 | 2380 | 1676 | 2165 | 2159 | 1036 | 2751 |
| Linkedspl | 61 869 | 44 196 | 30 | 0 | 163 639 | 163 639 | 3613 | 0 | 37 807 | 1134 | 2131 | 2140 | 0 | 0 |
| OMIM | 104 540 | 0 | 0 | 31 468 | 137 682 | 0 | 0 | 137 682 | 0 | 0 | 2118 | 2120 | 0 | 0 |
| Pharmgkb | 6543 | 868 | 1442 | 486 | 33 508 | 23 322 | 17 610 | 25 460 | 1643 | 1638 | 2131 | 2140 | 1376 | 2208 |
| Pharmgkb-offside | 450 230 | 1332 | 0 | 10 097 | 877 604 | 438 802 | 0 | 438 802 | 7149 | 6138 | 0 | 0 | 50 905 | 52 159 |
| Sider | 2598 | 893 | 0 | 1705 | 68 424 | 68 424 | 0 | 68 424 | 858 | 868 | 0 | 0 | 814 | 1428 |
| STRING | 19 385 | 0 | 19 385 | 0 | 5 969 250 | 0 | 5 969 250 | 0 | 0 | 0 | 2131 | 2133 | 0 | 0 |
| Total | 971 874 | 59 322 | 95 280 | 49 253 | 8 530 037 | 817 110 | 7 199 428 | 975 011 | 51 146 | 6250 | 15 917 | 2838 | 54 576 | 52 187 |
| (b) Evaluation tasks | ||||||||||||||
| Test | Purpose | Validation | Drug–Target Space | # Task | # Avg train pairs | # Avg drugs for training | # Avg targets for training | # Avg test pairs | # Avg test positive | # Avg test negative | # Avg drugs for testing | # Avg targets for testing | ||
| 0 | General | k-fold cross internal validation (k = 10) | All | 10 | 11 016 | 5337 | 3087 | 1483 | 676 | 807 | 1190 | 1039 | ||
| 1 | Screening with connectivity spaces (e.g. isolated drugs or targets) | k-fold cross internal validation (k = 10) | Connectivity space | 90 | 11 769 | 5654 | 3221 | 929 | 471 | 458 | 637 | 551 | ||
| 2 | Target screening when drugs are within/beyond category | k-fold cross internal validation (k = 3) + Drug categories | Category space (drugbank category) | 36 | 12 164 | 5731 | 3252 | 152 | 76 | 76 | 32 | 73 | ||
| Category space (linkpl class) | 36 | 12 108 | 5732 | 3201 | 263 | 132 | 132 | 32 | 192 | |||||
| 3 | Drug screening when targets are within/beyond category | k-fold cross internal validation (k = 3) + Protein categories | Category space (family) | 36 | 12 045 | 5721 | 3252 | 389 | 195 | 195 | 109 | 120 | ||
| Category space (protein class) | 36 | 11 965 | 5685 | 3247 | 549 | 275 | 275 | 165 | 194 | |||||
| 4 | Searching for a specific drug/target | Internal validation with m search (n = 10) | All targets (500) | 10 | 12 207 | 5735 | 3239 | 534 | 33 | 501 | 1 | 534 | ||
| Internal validation with m search (n = 10) | All drugs (500) | 10 | 12 196 | 5717 | 3253 | 545 | 44 | 501 | 545 | 1 | ||||
| 5 | Drug repurposing for a specific disease | Version-based external validation + Disease specific | Category space (10 disease) | 40 | 12 240 | 5735 | 3253 | 44 | 22 | 22 | 24 | 14 | ||
| 6 | Clinical trial-based external validation + Disease-specific | Category space (10 disease) | 40 | 12 240 | 5735 | 3253 | 25 | 13 | 13 | 10 | 11 | |||
For the evaluation component, we designed seven main Tests (344 Tasks in total) based on the Perspectives (i.e. Perspectives of validation and data spaces) that were used for the generation of training and testing sets (see Table 1(b)). Specifically, Tests 0–4 (i.e. internally validated Jobs) generated existent associations (i.e. positives) based on internal validation, in which the random selection of the entire drug–target space was used to generate nonexistent (i.e. negative) testing associations for Test 0 (10 Tasks), connectivity spaces were used for Test 1 (90 Tasks), category spaces were used for Tests 2 and 3 (144 Tasks) and a search of drug–target space was used for Test 4 (20 Tasks). Tests 5–6 (i.e. external validated Jobs) generated positive associations based on external validation, in which versioning- and clinical trial-based validation was used to generate the positive training and testing sets for Tests 5 (40 Tasks) and 6 (40 Tasks), respectively, and disease-based categories were used to generate negative associations for testing.
![Organization of the proposed benchmark. The evaluation Tasks were generated based on a multipartite network and publicly available third-party resources (e.g. Drugbank [26] and ClinicalTrials.gov [39]). Two Perspectives, validation and data spaces, were used to generate the training and testing associations. Specifically, there were two types of validation: internal validation (e.g. K-fold cross-validation, drug category- and target category-based) and external validation (e.g. versioning- and clinical trial-based), used to generate the positive training and testing associations. Additionally, two types of data spaces, connectivity space (e.g. Scenario 1—default, 2—semicold start and 3—cold start) and category space [e.g. Test End Only (TEO), Train End Only (TRO), Test End Excluded (TEE) and Train End Excluded (TRE)], were designed to generate the negative testing associations. In total, seven main Tests comprising 344 Tasks were provided in the benchmark based on the two Perspectives, in which Test 0 was for general drug–target prediction (10 Tasks), Test 1 for screening for drug–target associations with connectivity spaces (90 Tasks), Tests 2–3 for target and drug screening with category space (144 Tasks), Test 4 for drug and target searching (20 Tasks) and Tests 5–6 for drug repurposing (40 Tasks).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/bib/23/4/10.1093_bib_bbac199/1/m_bbac199f1.jpeg?Expires=1750214628&Signature=YgCNA6chetgnfRG7cqXtoQewuyMDhfXX1mFJ1rlROg5-V17FqEWYHmbAWgNjDYsC~0Hiwsl06VHIJks376sef1wagP1UXBFRAJmqXuAoc8ANQ8Y3z6AWq-zEkMGbRDAPDHFMtEscuN6vPQPak-AgRGvrZPUnaBPHj1piF~ngxZMTPBjpcKPX7fRRcqfQg3RH5Cz317vyFXD-YgJpccOD15owtCT4VyqdtA1Qj0ffDc8wz9siU-AxqzAbaGUFHTCQvO0nRmxu0E1rFF9lh8qjc2PI1tKS13uU~WffZPqsKWuW~4hcFBVjGktqcTqe7wfeW0VGTg5HUWNpwJ-OAqrjXw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Organization of the proposed benchmark. The evaluation Tasks were generated based on a multipartite network and publicly available third-party resources (e.g. Drugbank [26] and ClinicalTrials.gov [39]). Two Perspectives, validation and data spaces, were used to generate the training and testing associations. Specifically, there were two types of validation: internal validation (e.g. K-fold cross-validation, drug category- and target category-based) and external validation (e.g. versioning- and clinical trial-based), used to generate the positive training and testing associations. Additionally, two types of data spaces, connectivity space (e.g. Scenario 1—default, 2—semicold start and 3—cold start) and category space [e.g. Test End Only (TEO), Train End Only (TRO), Test End Excluded (TEE) and Train End Excluded (TRE)], were designed to generate the negative testing associations. In total, seven main Tests comprising 344 Tasks were provided in the benchmark based on the two Perspectives, in which Test 0 was for general drug–target prediction (10 Tasks), Test 1 for screening for drug–target associations with connectivity spaces (90 Tasks), Tests 2–3 for target and drug screening with category space (144 Tasks), Test 4 for drug and target searching (20 Tasks) and Tests 5–6 for drug repurposing (40 Tasks).
Data collection and processing
To generate the full datasets, we collected the linked data version of the human disease network [31], diseasome. The Drugbank, GOA, iRefindex, KEGG [27], linkedspl [33], OMIM [35], Pharmgkb [36] and SIDER [37] were collected from Bio2rdf release 4 [40]. STRING V11 [38] was directly downloaded. To integrate the databases, we defined the common node spaces for drugs, targets and diseases, in which Drugbank drugs and targets were for drugs and targets, and OMIM was for diseases (see Supplementary Figure 1, see Supplementary Data available online at https://dbpia.nl.go.kr/bib). The common entity identifiers (IDs) were used for mapping. Specially, the identifiers from Unified Medical Language System (UMLS) [41], DBpedia and Wikipedia [42], KEGG, PubChem [43] and Pharmgkb are used for mapping in drug space; UniPort Knowledgebase [44], HUGO Gene Nomenclature Committee [45], GenAtlas [46] and OMIM were for target space, whereas DBpedia, UMLS and Systematized nomenclature of medicine clinical terms (SNOMED CT) [47] are for disease space. We utilized owl:sameAs to provide a mapping across different datasets and kept the original entities and the associations in each dataset without integrating similar concepts from different datasets into one data point (i.e. entity). We obtained the drug chemical structure formatted in the Simplified Molecular Input Line Entry System [48] and gene sequence from Drugbank. Targeting the drugs and targets from Drugbank in the common node spaces, we generated the drug–drug similarity and protein–protein similarity matrices based on the Tanimoto similarity with Chemistry Development Kit [49] and Smith–Waterman algorithm [50]. The quantitative values in other datasets were not incorporated or computed in the proposed benchmark as it is challenging to normalize the quantitative values across the different datasets for the computation. In practice, an Resource Description Framework (RDF) triple store, GraphDB [51], was adopted to manage the network.
![Logistics of two Perspectives (i.e. data spaces and validation) in the proposed benchmark. For the Perspective of data space, connectivity space- and category space-based Jobs were proposed to generate the nonexistent associations (i.e. negatives) associations for testing. Specifically, three Scenarios reflected the connectivity for nodes used for training, which were Scenario 3—cold start (i.e. a pair of nodes was isolated if the association was removed), Scenario 2—semicold start (i.e. one of the nodes in a pair was isolated if the association was removed) and Scenario 1—default (i.e. none of the nodes in a pair was isolated if the association was removed). There were nine Jobs for connectivity space-based Jobs, which included TT space, TC space, TA node space, CC space, CA node space, and AA node space for Scenario 1, SS and SU for Scenario 2 and DI in testing space for Scenario 3. Four Jobs were designed for category space-based Jobs, which were TEO, TRO, TEE, and TRE. For the Perspective of validation, both internal and external validation were used. Three Jobs were used for internal validation, which were k-fold cross-validation-based, drug category-based and target category-based Jobs. For a demonstration example, the heat maps in the two latter Jobs showed similarities among and between the different categories (e.g. dopamine antagonists and gaba modulators) and protein family (e.g. cell division protein kinase and sodium/chloride-dependent transporter). Two Jobs were used for external validation, which included versioning-based and clinical trial-based Jobs. For a demonstration example, six targets were allocated based on a query of breast cancers. The proteins phosphatidylinositol 4,5-bisphosphate 3kinase catalytic subunit alpha isoform, androgen receptor and cellular tumor antigen p53 were associated with the drugs adenosine triphosphate, diethylstilbestrol and triethyl phosphate based on validation with a newer version of Drugbank. The genes DNA repair protein RAD51 homolog 1 and serine/threonine-protein kinase Chk2 were associated with drugs fulvestrant, capecitabine and cyclophosphamide-based on validation with ClinicalTrials.gov. The structures of drugs and proteins were obtained from Drugbank and Protein Data Bank [59], respectively.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/bib/23/4/10.1093_bib_bbac199/1/m_bbac199f2.jpeg?Expires=1750214628&Signature=G3RiqC53bMQWIVm5I90tU9JlLQBnejHPgFczxc8E9vjCh5SbjpyyYrYhVB83FE4K~Vakt3~9-ex1hs09~dRTtCPGTatcu-LA15H0lD1tQM45gcQxTKUYwe1v2YgBZb6Y5Zp3xflr-6eC9nxJHH547ayWN8X2S5Rk0cROrac6Y9H3hqOWWEntlXlQ4LNfROpMl5mO91NL8E1ekurRsuSxZs49ymbBDOVkx-UcRsbC9ZyfY4LD0LWSS58CwVvqemgl2NfOXF6GdSgLUVi~ZsCCuxNIhycF9pEwkrUnnQX9FHWLfIXQ7EU46APQKdtZQAxRU28BgEhhwP2LvKYeaGd8ug__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Logistics of two Perspectives (i.e. data spaces and validation) in the proposed benchmark. For the Perspective of data space, connectivity space- and category space-based Jobs were proposed to generate the nonexistent associations (i.e. negatives) associations for testing. Specifically, three Scenarios reflected the connectivity for nodes used for training, which were Scenario 3—cold start (i.e. a pair of nodes was isolated if the association was removed), Scenario 2—semicold start (i.e. one of the nodes in a pair was isolated if the association was removed) and Scenario 1—default (i.e. none of the nodes in a pair was isolated if the association was removed). There were nine Jobs for connectivity space-based Jobs, which included TT space, TC space, TA node space, CC space, CA node space, and AA node space for Scenario 1, SS and SU for Scenario 2 and DI in testing space for Scenario 3. Four Jobs were designed for category space-based Jobs, which were TEO, TRO, TEE, and TRE. For the Perspective of validation, both internal and external validation were used. Three Jobs were used for internal validation, which were k-fold cross-validation-based, drug category-based and target category-based Jobs. For a demonstration example, the heat maps in the two latter Jobs showed similarities among and between the different categories (e.g. dopamine antagonists and gaba modulators) and protein family (e.g. cell division protein kinase and sodium/chloride-dependent transporter). Two Jobs were used for external validation, which included versioning-based and clinical trial-based Jobs. For a demonstration example, six targets were allocated based on a query of breast cancers. The proteins phosphatidylinositol 4,5-bisphosphate 3kinase catalytic subunit alpha isoform, androgen receptor and cellular tumor antigen p53 were associated with the drugs adenosine triphosphate, diethylstilbestrol and triethyl phosphate based on validation with a newer version of Drugbank. The genes DNA repair protein RAD51 homolog 1 and serine/threonine-protein kinase Chk2 were associated with drugs fulvestrant, capecitabine and cyclophosphamide-based on validation with ClinicalTrials.gov. The structures of drugs and proteins were obtained from Drugbank and Protein Data Bank [59], respectively.
Benchmark design
In general, the purpose of the evaluation was to assess how well a model can predict drug–target associations by separating existent associations (i.e. positives) from a highly imbalanced large number of nonexistent associations (i.e. negatives). Conventionally, three characteristics of datasets were widely used for evaluations: (i) the connectivity pattern of the drugs and targets that underlie topological context and inherent connection profiles [21, 52, 53], (ii) the categories of drugs and targets in real scenarios [54–56] and (iii) the validation of the associations internally and externally [55, 57, 58]. As such, we designed the seven evaluation Tests that generated the training and testing associations based on the two Perspectives—validations and data spaces. It should be noted that to distinguish the hierarchical level of logic used for the evaluation tasks, we used Tests, Perspectives, Jobs, Scenarios and Tasks to represent the evaluation task in each logic layer, in which each child concept was considered to be the subtask of its respective parent (e.g. Perspective of validation contains internal validation-based Jobs and external validation-based Jobs).
Perspective of validation
Two types of validations, consisting of both internal and external validation-based Jobs, were designed to generate positive training and testing associations.
Internal validation-based jobs
Three validations were used: (i) k-fold cross validation-based, in which the original drug–target associations were randomly partitioned into k equal-sized subsamples (without resulting in any isolated nodes in the network being built by any of the remaining K-1 subsamples), and then included in repeated k independent experiments conducted using each subsample for testing (i.e. positives testing set) and the remaining k – 1 subsamples for training (i.e. positives training set) [4]; (ii) drug category-based, in which the drug–target associations were partitioned for training and testing, included pairs with similar drugs from two different category systems for drugs, drug categories from Drugbank and pharmacologic class from DailyMed [60] and (iii) target category-based, where the drug–target associations were partitioned for training and testing based on different category systems for targets, family and protein class from Panther [61].
External validation-based jobs
Two validations were used to evaluate the algorithms’ ability in predicting new associations that existed in the newer version of training data and recently conducted clinical trials: (i) versioning-based, in which novel drug–target associations in the latest version of Drugbank were tested with the older version used as the training data and (ii) clinical trial-based, in which novel drug–target associations obtained from the latest clinical trials at ClinicalTrials.gov were tested based on the entire datasets in the benchmark used as the training data. Specifically, for a particular target of a disease, the drugs tested for clinical trial interventions under ‘recruiting’ status were considered a novel drug–target pair for testing. For the two external validation, the targets were categorized by the associated diseases.
Perspective of data space
Two types of data spaces were designed to generate negative drug–target associations for testing: topological structures in the drug–target space and in the node category space.
Connectivity space-based jobs
The permutations of connectiveness levels between the nodes supplied to an algorithm for predictive purposes were tested in the evaluation. Specifically, the nodes associated with the drug–target associations were classified into three spaces: (i) Test Node Space (TNS), (ii) Connected Node Space (CNS) and (iii) All Node Space (ANS). TNS consisted of all the drug and target nodes used for testing. CNS consisted of all drug and target nodes with a drug target association existing between them. ANS consisted of all drug and target nodes. Consequently, nine types of negative associations were designed based on three Scenarios, which reflected the connectivity of drugs and targets. Scenario 1 (default) was defined as ‘given a pair of drug and target, neither of the nodes (a drug or target) are isolated if the association is removed for testing’. In Scenario 1, testing nodes were those nodes in the CNS. Six Jobs can be generated, which included Test–Test space-based (TT, a pair of nodes both coming from TNS), Test-Connected space-based (TC, a pair of nodes coming from TNS and CNS), Test-All node space-based (TA, a pair of nodes coming from TNS and ANS), Connected-Connected space-based (CC, a pair of nodes both coming from CNS), Connected-All node space-based (CA, a pair of nodes coming from CNS and ANS) and All-All node space-based (AA, a pair of nodes both coming from ANS). Scenario 2 (semi-cold start) was defined as ‘given a pair of drug and target, one of the two nodes (either a drug or target) is isolated if the association is removed for testing’. In Scenario 2, one of the nodes was not in the CNS. Based on ‘guilt-by-association’ [4, 52], the two nodes were considered similar if they connected to a common node. Therefore, two types of negative associations can be generated for testing, which were Semi-isolated with Similar nodes (SS) and Semi-isolated with Unsimilar nodes (SU). Scenario 3 (cold start) was defined as ‘given a pair of drug and target, both nodes (drug and target) are isolated if the association is removed for testing’. In Scenario 3, both nodes were not in the CNS; accordingly, a pair of Double Isolated (DI) nodes can be created for negative associations.
Category space-based jobs
Drug-target associations were selected for testing similar/ dissimilar drugs or targets based on a category. Four types of negative associations were designed as follows: (i) TEO, in which testing negative pairs (a source node and an end node) were generated based on a source node of a testing positive and similar end nodes to the end node of testing positive in a category, (ii) TRO, in which testing negatives were generated based on a source node of testing positives and similar end nodes to the end node of testing positive in a category, (iii) TEE, in which testing negative pairs were generated based on a source node of a testing positive and nonsimilar end nodes to the end node of testing positive beyond a category and (iv) TRE, in which testing negatives were generated based on a source node of nontesting positives and nonsimilar end nodes to the end node of testing positive beyond a category.
Evaluation tasks
Based on the combinations of the Perspectives of data space and validation, seven evaluation Tests were generated, in which the positive associations for training and testing were generated based on the validation-based Perspective, and the negative associations for testing were generated based on the data space-based Perspective. Please note that the negative associations for training were not provided in the benchmark Tasks as those associations can be generated to improve the performances in different algorithms [21, 62, 63]. To conduct a fair comparison, we removed the drugs, targets and the corresponding drug–target associations from the evaluation tasks when the drugs and targets did not have chemical structure and gene sequence information in the benchmark as those entities and associations cannot be processed by the structure- and sequence-based methods.
To organize the Tasks, we classified them as follows.
Internally validated tests
Test 0 (10 Tasks): This Test was designed to conduct a general evaluation of the drug–target prediction. The k-fold cross-validation was used to generate the positive training and testing pairs. The negative testing pairs were randomly selected. In practice, k was set to 10.
Test 1 (90 Tasks): This Test was designed to evaluate the drug–target prediction when drugs and targets were at different connectivity spaces (e.g. isolated drugs or targets). The k-fold cross-validation was used to generate the positive training and testing pairs. Nine different connectivity spaces were used for the selection of negative testing pairs. k was set to 10.
Test 2 (72 Tasks): This Test was designed to evaluate the drug–target prediction when drugs were within or beyond two categories (i.e. drug categories from Drugbank and pharmacologic class from DailyMed [60]). The k-fold cross-validation was used to generate the positive training and testing pairs. Four kinds of category spaces for drugs were used for the selection of negative testing pairs. k was set to 3.
Test 3 (72 Tasks): This Test task was designed to evaluate the drug–target prediction when targets were within or beyond two categories (i.e. family and protein class from Panther [61]). The k-fold cross-validation was used to generate the positive training and testing pairs. Four kinds of category spaces for targets were used for the selection of negative testing pairs. k was set to 3.
Test 4 (20 Tasks): This Test was designed to evaluate the searching for drugs or targets when a target or a drug is given. N searches were conducted with a specified search space (e.g. 500 drugs or targets). In practice, N was set to 10.
Externally validated tests
Test 5 (40 Tasks): This Test was designed to evaluate the drug–target prediction for M diseases. A versioning-based validation was used to generate the positive testing pairs with the whole benchmark data that was used as positive training pairs. Four kinds of category spaces for targets were used for the selection of negative testing pairs. In practice, M was set to 10.
Test 6 (40 Tasks): This Test was designed to evaluate the drug–target prediction for M diseases. A clinical trial-based validation was used to generate the positive testing pairs with the whole benchmark data that was used as positive training pairs. Four kinds of category spaces for targets were used for the selection of negative testing pairs. M was set to 10.
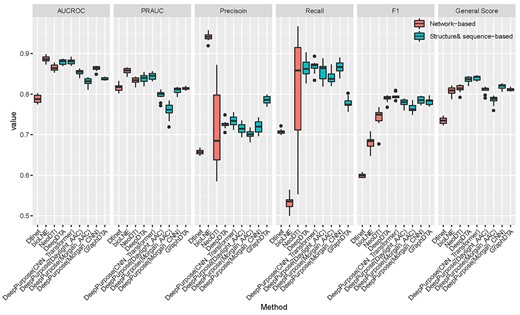
Results of six methods for Test 0 for all the evaluated methods.
Predictive models in evaluation
Based on the data sources used as the input, two types of algorithms were used: network-based methods and structure- and sequence-based methods: (i) Network-based methods are the methods that used any graphical information from the proposed benchmark as the input, which includes multiple types of biomedical entities, such as drugs, targets, diseases, side effects and pathways, and the corresponding information from multipartite (including drug–target bipartite) networks. In practice, we used three state-of-the-art network-based methods: DTINet [21], Bio-Linked Network Embeddings (bioLNE) [64] and NEural integration of neighbOr information for DTI prediction (NeoDTI) [65]. For DTINet and NeoDTI, we used drug–target, drug–disease, protein–disease, drug–side effect, protein–protein, drug–drug interaction as well as drug–drug similarity, and protein–protein similarity matrices as the input data. For bioLNE, we used drug, target, disease, side effect, chromosomal location, drug category, drug group, drug substance, food, module, pathway, variant location, haplotype, disease feature and disease symbol-related assertions. (ii) Structure- and sequence-based methods are the methods that primarily used the chemical structure of drugs and sequence of proteins as the input. The drug chemical structure and gene sequence were collected from Drugbank. In practice, we considered that DeepPurpose [66], DeepDTA [67] and GraphDTA [68] are state-of-the-art methods. For DeepPurpose, we used diverse encoding methods with the default setting, such as Convolutional Neural Network (CNN), Transformer, Daylight, AAC and Morgan for the structure and sequence information. For DeepDTA, we implemented it based on the DeepPurpose framework, in which structure and sequence information were both encoded with CNN. For NeoDTI, the parameter ranges for the grid search specified as dimension of node embedding|$d=\big\{256,512,1024\big\}$|, dimension of the projection matrices |$k=\big\{256,512,1024\big\}$|, repetition time of neighborhood information aggregation |$p=\big\{1,2,3\big\}$|. For bioLNE, the parameter ranges for the grid search specified as classification {J48, SVM, Random Forest, Logistic Regress}, Binary Operator {average, hadamard, wrighted-L1 and L2}, weights for DBSI and classification are {0.0–0.9} with 0.1 increment and default performed parameters of Node2Vec [69]. For GraphDTA and DTINet, the default parameters were used.
Evaluation metrics
Three metrics were mainly used to assess the quality of the predictions, including area under the receiver operating characteristic curve (AUC ROC) [56, 57], Precision/Recall (PR AUC) and F1 measure (including Precision and Recall). In practice, we normalized the three metrics |$Mean\ \big( \mbox{Area Under the Curve Receiver Operator} \mbox{Characteristic} \ (AUCROC) + \mbox{Precision Recall Area Under the} \mbox{Curve} \ (PRAUC)+F1\big)$| to obtain a balanced score to better identify the best-performing method in general. In addition, we also provide Precision, Recall and F1 measures at top k and mean average precision (MAP) at top k search results for Test 4. The AUC ROC, PR AUC and F1 scores were calculated by the ROC JAVA library (https://github.com/kboyd/Roc), the Weka evaluation package [58] and scikit-learn package [70].
Evaluation Results
Existing methods were categorized into two distinct categories for the purpose of evaluating our benchmark based on the input data used: (i) network-based and (ii) structure- and sequence-based methods. For network-based methods, DTINet [21], bioLNE [64] and NeoDTI [65] are considered state-of-the-art for comparison. For structure- and sequence-based, DeepPurpose [66], DeepDTA [67] and GraphDTA [68] were adopted. We conducted a general evaluation (Test 0) to select the best-performing methods as a representative for each category and showed their results for the rest Tasks (Tests 1–6) in the main manuscript. The complete results of all the experiments for all the evaluated methods are shown in the supplements.
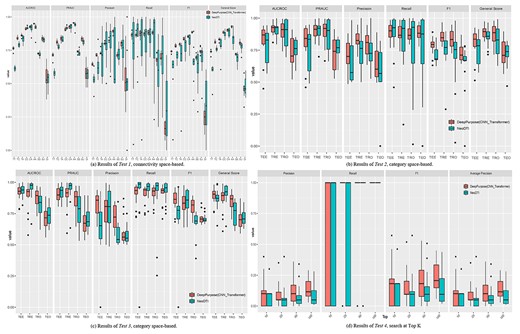
Results of internally validated Tests (Tests 1–4) for DeepPurpose and NeoDTI.
Our results (see Figure 3) show, in general, structure- and sequence-based methods performed better than network-based methods (average AUCROC: 85.72 versus 84.67%, PRAUC: 81.01 versus 83.55%, Precision: 73.00 versus 77.07%, Recall: 84.52 versus 68.24%, F1: 78.23 versus 67.35%, general score: 81.65 versus 78.52%). The best methods for network-based and structure- and sequence-based methods are DeepPurpose (CNN and Transformer) (average AUCROC: 88.01%, PRAUC: 84.44%, Precision: 73.40%, Recall: 86.73%, F1: 79.49%, general score: 83.98%) and NeoDTI (average AUCROC: 86.52%, PRAUC: 83.32%, Precision: 71.40%, Recall: 80.78%, F1: 74.19%, general score: 81.34%). For each metric, we observed that bioLNE performed the best in terms of AUCROC (88.65%), PRAUC (85.73%) and Precision (94.09%) whereas DeepPurpose (CNN and Transformer) performed the best in terms of Recall (86.73%) and F1 (79.49%). Notably, compared with other methods, NeoDTI had a large standard deviation for Precision (9.20%) and recall (13.05%). For the following Tasks, we considered NeoDTI and DeepPurpose (CNN and Transformer) named DeepPurpose for short as two representative methods and show their results here (the results for the rest methods are shown in Supplementary Material).
Internally validated tests
We conducted Tests 1–3, which were internally validated Tests. Regarding Test 1, in general, DeepPurpose (average general score: 78.67%) outperformed NeoDTI (74.09%) in all six Jobs across all 90 Tasks constituting the connectivity space-based Jobs (see Figure 4A). For Scenario 1—Default (i.e. no isolation of nodes resulted if an association is removed), the performance increased when the negatives are sampled from a broader connective space, and achieved the best for type AA (e.g. general score of DeepPurpose: 93.00% versus NeoDTI: 83.57% for AA). According to the ‘guilt-by association’ principle, the nodes were more similar when sharing more common neighbors [71]. The tested methods performed excellently when the positive and negative pairs were topologically distinct (e.g. AA) but performed much worse when they were more topologically connected (e.g. general score of DeepPurpose: 73.76% versus NeoDTI: 59.12% for TT).
For the cold start problem, methods performed the best in Scenario 1—default (AUCROC: 85.30%, PRAUC: 84.31%, Precision: 72.91%, Recall: 84.15%, F1: 75.98%, general score: 81.86%), and then Scenario 2—semicold start (AUCROC: 77.75%, PRAUC: 77.87%, Precision: 74.48%, Recall: 66.70%, F1: 68.38%, general score: 74.67%), in which isolation of one node resulted if an association was removed and finally Scenario 3—cold start (AUCROC: 54.29%, PRAUC: 57.20%, Precision: 53.28%, Recall: 24.24%, F1: 29.21%, general score: 46.90%), in which the isolation of both nodes resulted if the association was removed. The failure of the cold start problem in prediction indicated the necessity of predictive models for new drugs and new targets. Particularly, we learned that, compared with other jobs, DI has large standard deviations for other evaluated methods as shown in Supplementary Figure 2 (see Supplementary Data available online at https://dbpia.nl.go.kr/bib) (e.g. AUCROC: 8.11%, PRAUC: 6.96%, Precision: 23.04%, Recall: 21.00%, F1: 17.69%, general score: 9.36%).
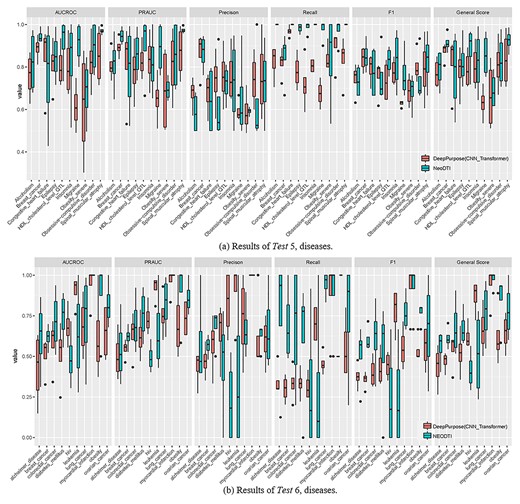
Results of externally validated Tests (Tests 5–6) for DeepPurpose and NeoDTI.
Regarding Tests 2 and 3, in general, DeepPurpose (average general score: 82.69% for drug category-based Jobs and 83.05% for target category-based Jobs) outperformed NeoDTI (78.83% for drug category-based Jobs and 81.47% for target category-based Jobs) across all four category spaces and four categories covering the 144 Tasks (see Figure 4B, C). Although DeepPurpose was selected as a representative model, bioLNE (83.84% for drug category-based Jobs and 84.46% for target category-based Jobs) was the best performing model (see Supplementary Tables 1 and 2, see Supplementary Data available online at https://dbpia.nl.go.kr/bib). Regarding the category space-based Jobs, TRE is the best performing (85.16% for drug category-based Jobs and 87.49% for target category-based Jobs), and TEO is the worst performing (73.72% for drug category-based Jobs and 71.11% for target category-based Jobs), which was consistent with the design of the evaluation—it was more difficult to separate the positive and negative drug–target pairs if the drugs and targets were similar in the two pairs. We also noticed that different categories had similar prediction results for drug (e.g. ‘drugbank category’: 78.82% and ‘linkpl class’: 78.84% for NeoDTI, ‘drugbank category’: 81.42% and ‘linkpl class’: 83.96% for DeepPurpose) and target categories (e.g. ‘family’: 81.33% and ‘protein class’: 81.60% for NeoDTI, ‘family’: 83.50% and ‘protein class’: 82.59% for DeepPurpose). The complete results for category-based and connectivity space-based Jobs of all the evaluated methods can be found in Supplementary Tables 1 and 2 (see Supplementary Data available online at https://dbpia.nl.go.kr/bib).
For Test 4 (see Figure 4D), DeepPurpose outperformed NeoDTI (MAP@10: 8.57% for DeepPurpose versus 4.86% for NeoDTI, MAP@20: 9.96% versus 5.06%, MAP@50: 11.13% versus 6.32%, MAP@100: 12.26% versus 7.10%). Among all the methods (see Supplementary Figure 3, see Supplementary Data available online at https://dbpia.nl.go.kr/bib), we noticed that bioLNE performed the best (MAP@10: 23.37%, MAP@20: 53.68%, MAP@50: 28.30%, MAP@100: 16.76%). Regarding the queries, we learned that the target queries performed better than drugs (MAP@10: 11.35% for targets versus 4.72% for drugs, MAP@20: 21.26% versus 14.87, MAP@50: 17.78% versus 9.62%, MAP@100: 16.72% versus 7.75%). Test 4 also suggested that the best k can be found among the top 20 to top 50 (MAP@20: 18.07% and MAP@50: 13.70%).
Externally validated tests
We conducted two types of externally validated Tests based on versioning (Test 5) and trials (Test 6). Regarding Test 5, despite the great performance achieved by DeepPurpose in the previous Tasks, NeoDTI (average general score: 83.10%) achieved slightly better performance compared with DeepPurpose (average general score: 77.79%) across the 40 Tasks constituting the versioning-based Jobs. Similar to the internal validation conducted, TRE performed the best (92.81% for NeoDTI and 82.98% for DeepPurpose), and TEO performed worst comparatively (74.24% for NeoDTI and 71.34% for DeepPurpose) (see Supplementary Figure 4A, see Supplementary Data available online at https://dbpia.nl.go.kr/bib).
Among the 10 diseases of interest, breast cancer had the best predictive performance (92.56% for NeoDTI and 90.59% for DeepPurpose) and obesity had the worst predictive performance (69.32% for NeoDTI and 71.63% for DeepPurpose). There are four diseases for which both NeoDTI and DeepPurpose performed well: breast cancer (91.41% for NeoDTI and 87.32% for DeepPurpose), spinal muscular atrophy (92.56% for NeoDTI and 83.70% for DeepPurpose), obsessive–compulsive disorder (83.01% for NeoDTI and 81.13% for DeepPurpose), and insomnia (82.50% for NeoDTI and 80.23% for DeepPurpose). Moreover, the results show that NeoDTI (88.2% for HDL_cholesterol_level_QTL and 85.61% for migraine) could be a good complementary tool for DeepPurpose.
Regarding Test 6, the performance of DeepPurpose and NeoDTI was worse than the previous Tasks, in which NeoDTI (average general score: 68.24%) outperformed DeepPurpose (average General Score: 63.29%) across the 40 Tasks constituting the trial-based Jobs. Similarly, among the four types, TRE performed the best (77.08% for NeoDTI and 63.38% for DeepPurpose) whereas TEO performed the worst (59.44% for NeoDTI and 57.53% for DeepPurpose) (see Supplementary Figure 4B, see Supplementary Data available online at https://dbpia.nl.go.kr/bib).
Among the 10 diseases, myocardial infarction had the best predictive performance (average general score: 97.2% for NeoDTI and average general score: 93.75% for DeepPurpose) whereas Alzheimer’s disease had the worst predictive performance (55.80% for NeoDTI and 43.67% for DeepPurpose). We also learned that NeoDTI (e.g. 84.58% for obesity) could be a good complementary tool for DeepPurpose, and vice versa for DeepPurpose (e.g. 86.77% for leukemia) in some cases. For the details of other methods for Tests 5 and 6, please refer to Supplementary Figure 5 (see Supplementary Data available online at https://dbpia.nl.go.kr/bib).
Analysis of failures in prediction
Investigating the prediction for Tests 5 and 6 helped identify situations in which existing methods could not perform well. We chose the best-performing (spinal muscular atrophy) and worst-performing disease (obesity) for NeoDTI in Test 5. We found that although NeoDTI successfully distinguished the positives from negatives in training data, it lacked the ability to predict when the targets were within the category (e.g. TEO and TRO). The results show NeoDTI tends to predict all the associations related to a target as a positive or a negative, which may result in a good performance when all the labels are all in positives or negatives (see be0000132 in TEO for spinal muscular atrophy in Figure 6A). However, it may result in bad performance if both positives and negatives are in testing pairs (see be0000215 in TEO and TRO for obesity in Figure 6B). Although NeoDTI performed well for some diseases, we found it cannot properly repurpose drugs for a given protein despite having the potential to predict the protein with a given drug (see TEE and TRE). A similar observation of myocardial infarction (best performing) versus HIV (worst) for a trial-based Job (Test 6) can be found in Supplementary Figure 6 (see Supplementary Data available online at https://dbpia.nl.go.kr/bib).
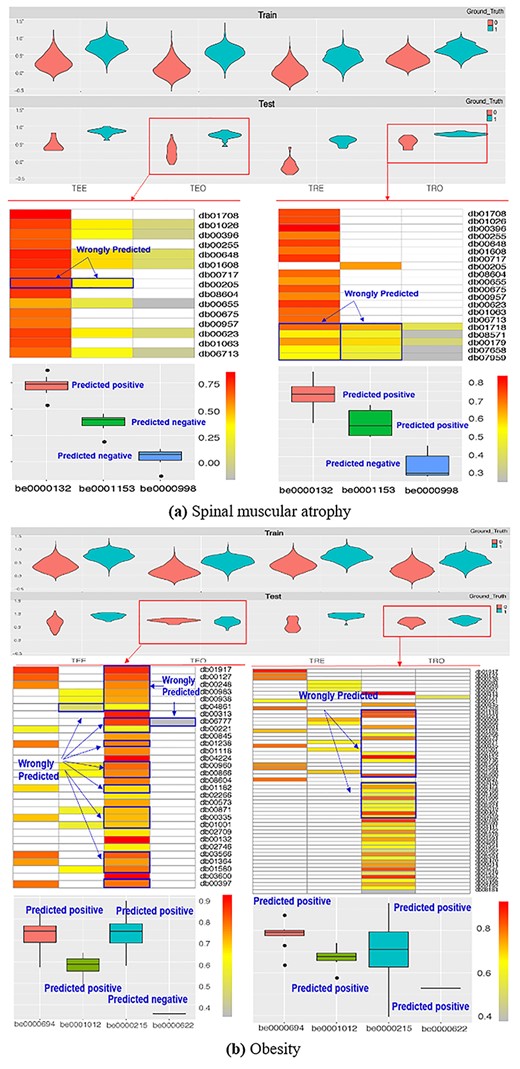
Best- and worst-performing predictions in Test 5 for NeoDTI.
Discussion
To design the benchmark, we extracted various biomedical associations from existing publicly accessible databases and knowledge bases and designed various evaluation Tasks to evaluate the ability of the predictive models to separate the positive and negative drug-target associations with similar drugs or targets defined for different purposes. We hope our work will provide a standardized and comprehensive way to evaluate the existing models as well as substantial information (e.g. features and associations of/among biomedical entities) to facilitate the selection of the most suitable predictive models in the real-world developmental process—the prediction of drug–target associations with a given a pair of drugs and targets, so as to lay the foundation necessary for the successful development of robust computational drug–target prediction methods similar to how the Text RetriEval Conference [72] has contributed to information retrieval and ImageNet [73] has contributed to visual recognition. Please note that our benchmark is designed for the methods requiring a pair of inputs and is not applicable to the methods similar to (Q)SAR [74] where only chemical structures are required.
The main advantages of our proposed benchmark include the creation of (i) a large-scale multidimensional network for prediction consisting of an extensive set of biomedical entities and diverse types of associations, and (ii) diverse prediction use-cases in which drugs and targets utilized for training and testing can be selected based on the difference of topological connectivity (connected versus isolated) or biomedical categories (e.g. drug category versus protein family). The datasets and evaluation Tasks are provided as off-the-shelf methods that can be easily adopted as a well-documented resource for drug development activities and scientific experiments. Although we have incorporated 11 repositories, more endeavors are needed to incorporate important repositories into the benchmark. The addition of datasets to our benchmark can be simply implemented by mapping to the common node space (i.e. the drugs targets from Drugbank and the diseases from OMIM). Currently, we are planning to incorporate more repositories (e.g. SymMap [75]) when their data are publicly accessible (see https://github.com/bioIKEA/IKEA_BETA_Benchmark/tree/master/data_space/output/datasets/pending for the pending repositories).
With our experiment, we found two limitations/biases of existing state-of-the-art methods. Firstly, they are incapable of handling new drugs and new targets (e.g. the poor performance of SS, SU and DI for Test 1) and secondly, they are incapable of properly repurposing drugs for a given protein—refer to be0000215 for obesity. As the discovery of the target-defined antineoplastic compounds is considered a more effective way for drug development (e.g. in cancers [76]), it drives researchers to develop novel repurposing methods to fill these needs. The proposed benchmark will provide a fine assessment of the effectiveness of the drug repurposing methods. Secondly, both DeepPurpose [66] and NeoDTI [65] perform worse when the training and testing nodes sharing more connections (e.g. the performance of CC > TA > TC > TT; SU > SS > DI). Although it was expected for a network-based method, such as NeoDTI, to be affected by the connectivity of the drugs and targets, it is a novel bias found for DeepPurpose. It, in a sense, demonstrated an indirect connection between topology-, structure- and sequence-based features for drugs and proteins in drug development contexts [28, 77, 78]. Although we addressed the bias caused by characteristics of data (e.g. topological structure), some biases in the practice of drug development (e.g. investigative bias) were not addressed. More evaluation Tasks are needed to perfect the investigation of the biases.
The evaluation Tasks in the benchmark have relied on the drug–target associations provided in Drugbank, which is considered as a ground truth. The drug–target associations in Drugbank were collected, curated and validated with multiple sources (e.g. PubMed, Therapeutic Target Database (TTD), Food and Drug Administration (FDA) labels, RxList, PharmGKB, textbooks) [26] and were used widely in drug–target prediction tasks. In our study, we simply trusted the existing knowledge bases to build our benchmark. Although we consider our contribution as the development of the benchmark, it is also important to keep updated on the associations that are trustworthy and supported by the experimental screen. On the other hand, the proposed benchmark was designed to provide heterogeneous biomedical information despite recognizing that it is a challenge for such information to be processed. To represent the knowledge graph, we used an undirected multidimensional network (i.e. edge types can be various but multiple and directed edges between two nodes are not allowed). We only kept one theme for each repository (e.g. only one type of association between the nodes), and used owl:sameAs to map similar concepts between the different repositories such that they can be linked with differing associations (e.g. reversed or conflict associations). For example, through the mappings between the disease entity ‘myocardial infarction’ (Diseasome: 3281) in Diseasome to the Omim (Omim:608446), and the side effect entity in Sider (Sider: C0027051) to the Omim (Omim:608446), drug–disease associations and drug–side effects associations can be utilized simultaneously. In our study, apart from the drug–target associations, the evaluation only covers the usage of partial associations (e.g. drug–drug and gene–gene similarity, drug–side effect, drug–drug and protein–protein interaction, drug–disease and protein–disease used in NeoDTI). The current benchmark is to assess the performance of drug–target prediction, and the heterogeneous datasets are designed to facilitate the computational but are not requisite. The usage of the other associations needed to be considered in the construction of the benchmark (e.g. the effect of removal of associations for network-based methods) or other prediction tasks (e.g. prediction of protein–protein interaction).
Our study only incorporated two types of methods: structure- and sequence-based and network-based methods, in our evaluation, as they utilized two distinct forms of input data—graphical structures of biomedical knowledge (including drug-target associations) and chemical structure and gene sequences. The two types were considered ideal representatives to demonstrate how our benchmark can provide input data to facilitate the computation. Although other types (e.g. matrix factorization, similarity/distance-based, feature-based and hybrid methods [14]) could not be tested in this study, they can be swapped as needed for individual evaluation use cases so long as the data to be evaluated on can be made into one of the two evaluated forms. The selection of six methods evaluated in our benchmark is based on three criteria: (i) methods of drug–target prediction published in the top-tier journals for computational biology from 2017; (ii) methods with open access source code and (iii) methods that are feasible for processing large training data. Although there are recently published excellent works (e.g. CoVex [79], a Bayesian ANalysis to determine Drug Interaction Targets (BANDIT) [80] and network-based proximity [54]), they cannot be implemented/included in the experiments due to the limited resources. Regarding the evaluation metrics, both AUCROC and PRAUC were not cut-off line sensitive (i.e. a threshold score was used to separate the positive and negative pairs for a prediction), whereas the F1 measure was. A debate exists on how a prediction should be given in real practice (e.g. probability estimation versus binary decision): Although the probability estimation may give a researcher more flexibility to set up a cut-off line, it is also challenging to determine a suitable cut-off line in practice [14]. Depending on the users’ preferences, further customization of the benchmarking evaluation may be necessary. Although the proposed benchmark is only designed for evaluating the effectiveness of prediction, we recognize that efficiency is also critical for the selection of a suitable method in the real-world screening of a large number of drug–target pairs. In general, the evaluation Tasks are scalable for the evaluated methods since the running time scales linearly with the number of Tasks for each main Test (Supplementary Figure 7, see Supplementary Data available online at https://dbpia.nl.go.kr/bib). As each Task is independent, in practice, a parallel computation can be applied for each of the evaluated methods, allowing for indefinite horizontal scaling as the resources would permit. On the other hand, although we set 500 drugs or targets in our search tasks (Test 4), it requires a larger size of search space in practice. Therefore, stress testing [81] to assess how stable a method can complete screening on a full combination of a large number of drug and target sets is needed.
We have proposed BETA, a large-scale benchmark that enables a comprehensive evaluation of drug-target predictive models to facilitate a selection of computational strategies for drug and target prescreening.
BETA provides an extensive multipartite network that is consisted of 0.97 million biomedical concepts and 8.5 million associations, in addition to 62 million drug–drug and protein–protein similarities.
BETA provides evaluation strategies that reflect five purposes with a total of seven Tests with 344 Tasks across multiple sampling and validation strategies.
Six state-of-the-art methods covering two broad method types (chemical structure- and gene sequence- and network-based) were evaluated with the developed Tasks across multiple Jobs (screening with different levels of connectivity, target/drug screening when drugs/targets are within/beyond category and drug repurposing for a specific disease).
Availability
The proposed method is available, along with the data and source code, at the following URL: https://github.com/bioIKEA/IKEA_BETA_Benchmark.
Data availability
The data that support the findings of this study are available on request from the corresponding author upon reasonable request.
Code availability
The code for data integration and the benchmark generation is available at https://github.com/bioIKEA/IKEA_BETA_Benchmark.
Funding
National Institute of Health (NIH) NIGMS (K99GM135488).
Author Biographies
Nansu Zong is an assistant professor at the Department of Artificial Intelligence and Informatics Research, Mayo Clinic. He works on computational drug development based on knowledge base and deep learning algorithms.
Ning Li is a research fellow at the Center for Structural Biology (CSB) of the National Cancer Institute, NIH. He works on the structural and functional study of protein kinase A involving the method of X-ray crystallography and cryo-EM.
Andrew Wen is a bioinformatician from Mayo Clinic. He is interested in utilizing informatics tools to build diverse applications in the health care area. He is an expert on natural language processing (NLP).
Victoria Ngo is a postdoctoral research fellow at VA Palo Alto Health System and Stanford Health Policy. Ngo is a health informaticist, and her research focuses on health equity and the optimization of information technology to improve the delivery and coordination of care in the community.
Yue Yu is a bioinformatician from Mayo Clinic, who is mainly working in the medical data standardization field. Yu is also interested in using artificial intelligence methods to solve biomedical problems.
Ming Huang is an assistant professor in the Department of AI and Informatics at Mayo Clinic. He is an expert in topic modeling and deep learning.
Shaika Chowdhury is a research fellow with the Mayo Clinic AI & Informatics, who studies deep learning-based precision medicine. Chowdhury is interested in utilizing knowledge graphs to improve the performance of deep learning models.
Chao Jiang is a PhD student at Auburn University. He works on diverse deep learning models and is particularly focused on graph neural networks.
Sunyang Fu is a senior data science analyst and biomedical informatics researcher at the Mayo Clinic. His research focuses on (i) designing and validating NLP techniques for clinical information extraction, (ii) developing informatics frameworks and processes to accelerate the secondary use of electronic health records (EHRs) for clinical research and (iii) discovering EHR heterogeneity and information quality through quantitative and qualitative methods.
Richard Weinshilboum is a professor at the Department of Molecular Pharmacology and Experimental Therapeutics, Mayo Clinic. He studies pharmacogenomics—the role of inheritance and individual variation in DNA sequence or structure in drug response.
Guoqian Jiang is a professor at the Department of Artificial Intelligence and Informatics Research, Mayo Clinic. He researches biomedical terminologies and ontologies, data standards, common data elements and common data models for clinical studies.
Lawrence Hunter is a professor of pharmacology and computer science at the University of Colorado. He focuses on the knowledge-driven extraction of information from the primary biomedical literature, the semantic integration of knowledge resources in molecular biology and the use of knowledge in the analysis of high-throughput data.
Hongfang Liu is a professor at the Department of Artificial Intelligence and Informatics Research, Mayo Clinic. The primary research focus of Hongfang Liu is to facilitate the secondary use of clinical data for clinical and translational science research and health care delivery improvement using data science, artificial intelligence and informatics approaches.
Mayo Clinic is a charitable, nonprofit academic medical center that provides comprehensive patient care and education in clinical medicine and medical sciences as well as extensive programs in research. Mayo Clinic includes Mayo Medical School, Mayo Graduate School, Mayo School of Graduate Medical Education, Mayo School of Continuous Professional Development and Mayo School of Health Sciences.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}