




Abstract
Major histocompatibility complex (MHC) possesses important research value in the treatment of complex human diseases. A plethora of computational tools has been developed to predict MHC class I binders. Here, we comprehensively reviewed 27 up-to-date MHC I binding prediction tools developed over the last decade, thoroughly evaluating feature representation methods, prediction algorithms and model training strategies on a benchmark dataset from Immune Epitope Database. A common limitation was identified during the review that all existing tools can only handle a fixed peptide sequence length. To overcome this limitation, we developed a bilateral and variable long short-term memory (BVLSTM)-based approach, named BVLSTM-MHC. It is the first variable-length MHC class I binding predictor. In comparison to the 10 mainstream prediction tools on an independent validation dataset, BVLSTM-MHC achieved the best performance in six out of eight evaluated metrics. A web server based on the BVLSTM-MHC model was developed to enable accurate and efficient MHC class I binder prediction in human, mouse, macaque and chimpanzee.
Introduction
Major histocompatibility complex (MHC) is a set of genes coding for cell surface proteins essential for immune surveillance. Due to variation in function, molecular structure and distribution, MHC molecules are classified into MHC class I, MHC class II and MHC class III. MHC class I proteins are transported to the cell surface in almost all cells and they display an antigen to provide signals for cytotoxic T lymphocytes including cluster of differentiation (CD8+). Found on the special antigen-presenting immune cells including macrophages, dendritic cells and B cells, MHC class II proteins typically bind with CD4+ receptors on the helper T cells to clear exogenous antigens. MHC class III genes are interspaced with class I and II genes on the short arm of chromosome 6, but their proteins play different physiologic roles. Of the three MHC classes, class I received the most attention in biomedical research due to its universality. For example, reduction in MCH class I is associated with poor prognosis [1]. Recently, a study [2] showed that tumors can escape T-cell responses by losing MHC class I proteins. MHC genes are highly polymorphic, and a particular variant of an MHC gene/protein is usually termed an MHC allele. Each MHC class contains multiple alleles. For example, in MHC class I, groups of human leukocyte antigen (HLA) A, B and C are defined, and each group is composed of multiple alleles. To determine the binding between an antigenic peptide and an MHC protein, researchers usually use such gold standard methods as enzyme-linked immunosorbent spot, intracellular cytokine staining, competitive binding assays, the direct binding assay and the real-time kinetic binding assay. Beyond these experimental methods, computational algorithms can help with inferring the binding affinity between an MHC and a potential antigen. In particular, computational approaches can efficiently prioritize plausible candidates for an optimal study design, thus alleviating both time and financial burden of the major wet-lab validation experiments. Based on known MHC-binding sequences accumulated from decades of experimental validation efforts, around 30 MHC binding prediction tools have been developed to predict new binding sequences of MHC proteins. A majority of these tools [3–6] focused on MHC class I, because it has more specific properties than class II in most molecular processes, including cancer vaccines [7], cytosolic double-stranded DNA (dsDNA) in rat thyroid epithelial cells [8] and peptide fragments of the foreign protein [9].
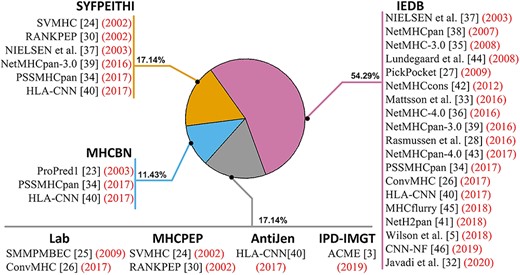
Usage frequency of major MHC binding databases to miscellaneous prediction tools.
Here, we review numerous MHC binding prediction tools and outline various aspects entailed in these computational approaches, including database sources, feature representation, machine learning strategies, etc. Our review is centered on MHC class I and it achieves a wide coverage by encompassing 13 MHC binding databases and 27 prediction tools. Moreover, we classified existing methods into two broad classes, machine learning and heuristic score. While reviewing the existing MHC class I binding tools, we identified a shared methodological limitation that all current approaches are limited to tackling a fixed peptide sequence length. To overcome this limitation, we developed a novel recurrent neural network solution based on bilateral and variable long short-term memory (BVLSTM), which achieves MHC class I prediction of variable peptide length. The implemented software product, with a specific application named BVLSTM-MHC. In the later portion of this review, we introduce the design of BVLSTM-MHC and demonstrate its superior performance in relation to existent MHC class I binding prediction tools.
Existing approaches
MHC binding databases
Experimentally verified MHC binding peptides were curated into databases. Over the last three decades, 13 MHC binding databases have been developed (Table 1). As of March 2021, nine databases are still functional, including two that had changed the hosting domain names. Two databases, Immune Epitope Database (IEDB) and Immuno Polymorphism Database of MHC (IPD-MHC), published updates within the prior 12 months. In terms of data quantity, the IEDB [10] hosts the largest collection of MHC-binding peptides with more than 900 000 peptides, followed by MHCBN [11] with 25 860 peptides. All existing databases focus on MHC class I and II binder, except for IMGT/MHC. And all databases mostly curate human data. These databases of experimentally verified MHC-binding sequences provide a foundation for the development of computational methods to predict an affinity score between a novel peptide and an MHC allele. Over the years, many MHC binding prediction tools have been developed based on data from these MHC binding databases. In following section, we focus on 27 mainstream prediction tools. The reliance on respective databases by various MHC class I binding prediction tools is illustrated in Figure 1. The largest database IEDB [10] has been used most frequently to MHC class I prediction tools, contributing to the development of 19 tools.
MHC binding databases
| Database | Address | Online | Last update | Entries | Species | Class |
|---|---|---|---|---|---|---|
| IEDB [10] | http://www.iedb.org/ | Yes | 2019 | >900,000 | Human, Mouse and Chimpanzee et al. | I, II |
| IPD-MHC [12] | https://www.ebi.ac.uk/ipd/mhc/ | Yes | 2019 | — | Canines, Felines et al. | I, II |
| IPD-IMGT/HLA [13] | https://www.ebi.ac.uk/ipd/imgt/hla/ | Yes | 2019 | — | Human | I, II |
| SYFPEITHI [14] | http://www.syfpeithi.de/ | Yes | 2012 | >7000 | Human | I, II |
| EPIMHC [15] | http://imed.med.ucm.es/epimhc/ | Yes | 2009 | 4875 | Human | I, II |
| MHCBN [11] | http://crdd.osdd.net/raghava/mhcbn/ | Yes | 2009 | 25,860 | Human, Mouse, rat et al. | I, II |
| AntiJen [16] | http://www.ddg-pharmfac.net/antijen/AntiJen/antijenhomepage.htm | Yes | 2005 | 15,454 | Human, Mouse, Rat et al | I, II |
| Bcipep [17] | https://webs.iiitd.edu.in/raghava/bcipep/ | Yes | 2005 | 1230 | Human, Mice et al | I |
| MPID-T [18] | http://variome.bic.nus.edu.sg/mpidt/index.html | Yes | 2005 | 187 | Human,Rat, Murine | I, II |
| MPID [19] | http://variome.bic.nus.edu.sg/mpid/ | No | 2003 | 86 | Uknown | I, II |
| JenPep [20] | http://www.jenner.ac.uk/JenPep | No | 2002 | 12,336 | Human | I, II |
| FIMM [21] | http://sdmc.krdl.org.sg:8080/fimm | No | 2002 | 1591 | Human | I |
| MHCpep [22] | http://wehih.wehi.edu.au/mhcpep/ftp. | No | 1994 | >4000 | Human, Mouse, rat, et al. | I, II |
| Database | Address | Online | Last update | Entries | Species | Class |
|---|---|---|---|---|---|---|
| IEDB [10] | http://www.iedb.org/ | Yes | 2019 | >900,000 | Human, Mouse and Chimpanzee et al. | I, II |
| IPD-MHC [12] | https://www.ebi.ac.uk/ipd/mhc/ | Yes | 2019 | — | Canines, Felines et al. | I, II |
| IPD-IMGT/HLA [13] | https://www.ebi.ac.uk/ipd/imgt/hla/ | Yes | 2019 | — | Human | I, II |
| SYFPEITHI [14] | http://www.syfpeithi.de/ | Yes | 2012 | >7000 | Human | I, II |
| EPIMHC [15] | http://imed.med.ucm.es/epimhc/ | Yes | 2009 | 4875 | Human | I, II |
| MHCBN [11] | http://crdd.osdd.net/raghava/mhcbn/ | Yes | 2009 | 25,860 | Human, Mouse, rat et al. | I, II |
| AntiJen [16] | http://www.ddg-pharmfac.net/antijen/AntiJen/antijenhomepage.htm | Yes | 2005 | 15,454 | Human, Mouse, Rat et al | I, II |
| Bcipep [17] | https://webs.iiitd.edu.in/raghava/bcipep/ | Yes | 2005 | 1230 | Human, Mice et al | I |
| MPID-T [18] | http://variome.bic.nus.edu.sg/mpidt/index.html | Yes | 2005 | 187 | Human,Rat, Murine | I, II |
| MPID [19] | http://variome.bic.nus.edu.sg/mpid/ | No | 2003 | 86 | Uknown | I, II |
| JenPep [20] | http://www.jenner.ac.uk/JenPep | No | 2002 | 12,336 | Human | I, II |
| FIMM [21] | http://sdmc.krdl.org.sg:8080/fimm | No | 2002 | 1591 | Human | I |
| MHCpep [22] | http://wehih.wehi.edu.au/mhcpep/ftp. | No | 1994 | >4000 | Human, Mouse, rat, et al. | I, II |
MHC binding databases
| Database | Address | Online | Last update | Entries | Species | Class |
|---|---|---|---|---|---|---|
| IEDB [10] | http://www.iedb.org/ | Yes | 2019 | >900,000 | Human, Mouse and Chimpanzee et al. | I, II |
| IPD-MHC [12] | https://www.ebi.ac.uk/ipd/mhc/ | Yes | 2019 | — | Canines, Felines et al. | I, II |
| IPD-IMGT/HLA [13] | https://www.ebi.ac.uk/ipd/imgt/hla/ | Yes | 2019 | — | Human | I, II |
| SYFPEITHI [14] | http://www.syfpeithi.de/ | Yes | 2012 | >7000 | Human | I, II |
| EPIMHC [15] | http://imed.med.ucm.es/epimhc/ | Yes | 2009 | 4875 | Human | I, II |
| MHCBN [11] | http://crdd.osdd.net/raghava/mhcbn/ | Yes | 2009 | 25,860 | Human, Mouse, rat et al. | I, II |
| AntiJen [16] | http://www.ddg-pharmfac.net/antijen/AntiJen/antijenhomepage.htm | Yes | 2005 | 15,454 | Human, Mouse, Rat et al | I, II |
| Bcipep [17] | https://webs.iiitd.edu.in/raghava/bcipep/ | Yes | 2005 | 1230 | Human, Mice et al | I |
| MPID-T [18] | http://variome.bic.nus.edu.sg/mpidt/index.html | Yes | 2005 | 187 | Human,Rat, Murine | I, II |
| MPID [19] | http://variome.bic.nus.edu.sg/mpid/ | No | 2003 | 86 | Uknown | I, II |
| JenPep [20] | http://www.jenner.ac.uk/JenPep | No | 2002 | 12,336 | Human | I, II |
| FIMM [21] | http://sdmc.krdl.org.sg:8080/fimm | No | 2002 | 1591 | Human | I |
| MHCpep [22] | http://wehih.wehi.edu.au/mhcpep/ftp. | No | 1994 | >4000 | Human, Mouse, rat, et al. | I, II |
| Database | Address | Online | Last update | Entries | Species | Class |
|---|---|---|---|---|---|---|
| IEDB [10] | http://www.iedb.org/ | Yes | 2019 | >900,000 | Human, Mouse and Chimpanzee et al. | I, II |
| IPD-MHC [12] | https://www.ebi.ac.uk/ipd/mhc/ | Yes | 2019 | — | Canines, Felines et al. | I, II |
| IPD-IMGT/HLA [13] | https://www.ebi.ac.uk/ipd/imgt/hla/ | Yes | 2019 | — | Human | I, II |
| SYFPEITHI [14] | http://www.syfpeithi.de/ | Yes | 2012 | >7000 | Human | I, II |
| EPIMHC [15] | http://imed.med.ucm.es/epimhc/ | Yes | 2009 | 4875 | Human | I, II |
| MHCBN [11] | http://crdd.osdd.net/raghava/mhcbn/ | Yes | 2009 | 25,860 | Human, Mouse, rat et al. | I, II |
| AntiJen [16] | http://www.ddg-pharmfac.net/antijen/AntiJen/antijenhomepage.htm | Yes | 2005 | 15,454 | Human, Mouse, Rat et al | I, II |
| Bcipep [17] | https://webs.iiitd.edu.in/raghava/bcipep/ | Yes | 2005 | 1230 | Human, Mice et al | I |
| MPID-T [18] | http://variome.bic.nus.edu.sg/mpidt/index.html | Yes | 2005 | 187 | Human,Rat, Murine | I, II |
| MPID [19] | http://variome.bic.nus.edu.sg/mpid/ | No | 2003 | 86 | Uknown | I, II |
| JenPep [20] | http://www.jenner.ac.uk/JenPep | No | 2002 | 12,336 | Human | I, II |
| FIMM [21] | http://sdmc.krdl.org.sg:8080/fimm | No | 2002 | 1591 | Human | I |
| MHCpep [22] | http://wehih.wehi.edu.au/mhcpep/ftp. | No | 1994 | >4000 | Human, Mouse, rat, et al. | I, II |
A summary of 27 MHC prediction tools
| Training strategy | Models | Dataset | Feature representationa | Prediction modelb | Evaluation criteriac | Code or Webserver |
|---|---|---|---|---|---|---|
| One-to-One | SYFPEITHI [14] | EMBL, RDBMS | constructing motif pattern | Heuristic score | None | http://www.syfpeithi.de/ |
| ProPred1 [23] | MHCBN | QM | Heuristic score | Sensitivity, Specificity, Accuracy, MCC | http://www.imtech.res.in/raghava/propred1 | |
| SVMHC [24] | MHCPEP, SYFPEITHI, ENSEMBL | sparse encoding | SVM | MCC, Specificity, Sensitivity | http://www.sbc.su.se/svmhc/ | |
| SMMPMBEC [25] | Laboratories | PMBEC | SMM | AUC | http://www.mhc-pathway.net/smmpmbec. | |
| ConvMHC [26] | IEDB, Sette and Buus laboratories | physicochemical scores | CNN | F1, Precision, Sensitivity | http://jumong.kaist.ac.kr:8080/convmhc | |
| PickPocket [27] | IEDB | PSSM | Heuristic score | AUC, PCC | None | |
| Rasmussen et al. [28] | IEDB | BLOSUM matrix | ANN | AUC, PCC | http://www.cbs.dtu.dk/services/NetMHCstabpan | |
| Bhasin et al. [29] | MHCBN, SWISS-PROT | QM, BIMAS | ANN | Sensitivity, Specificity, Accuracy, Precision | https://webs.iiitd.edu.in/raghava/nhlapred/neural.html | |
| RANKPEP [30] | MHCPEP, SYFPEITHI | PSSM | Heuristic score | www.mifoundation.org/Tools/rankpep.html | ||
| ForestMHC [31] | PRIDE, SysteMHC, Other | BLOSUM matrix, one-hot, physicochemical | RF | AUPR, Prec1% | None | |
| Javadi et al. [32] | IEDB | physicochemical, composition | RF | AUC, Accuracy, Sensitivity, PPV, Specificity, NPV | None | |
| Wilson et al. [5] | IEDB, MassIVE | biochemical | RF | AUC, Accuracy, Sensitivity, Specificity | None | |
| Mattsson et al. [33] | IEDB, SYSPETHI | BLOSUM matrix | ANN | PCC | None | |
| One-to-Many | PSSMHCpan [34] | MHCBN, IEDB, SYFPEITHI, Laboratories | BLOSUM matrix, PSSM | Heuristic score | ACC, AUC | https://github.com/BGI2016/PSSMHCpan |
| NetMHC-3.0 [35] | IEDB, SYFPEITHI | PSSMs | ANN | PCC, AUC | http://www.cbs.dtu.dk/services/NetMHC-3.0 | |
| NetMHC-4.0 [36] | IEDB, SYFPEITHI | BLOSUM matrix, sparse encoding | ANN | PCC, AUC | http://www.cbs.dtu.dk/services/NetMHC-4.0 | |
| NIELSEN et al. [37] | SYFPEITHI | sparse encoding, BLOSUM matrix | ANN | PCC, AUC, Sensitivity/PPV | None | |
| NetMHCpan [38] | IEDB | sparse encoding, BLOSUM matrix | ANN | Specificity, Sensitivity, PCC | http://www.cbs.dtu.dk/services/NetMHCpan/ | |
| NetMHCpan-3.0 [39] | IEDB, SYFPEITHI | BLOSUM matrix | ANN-based ML | PCC, AUC | www.cbs.dtu.dk/services/NetMHCpan-3.0 | |
| HLA-CNN [40] | MHCBN, IEDB, AntiJen, SYFPEITHI | HLA-Vec | CNN | SRCC, AUC | https://github.com/uci-cbcl/HLA-bind. | |
| NetH2pan [41] | IEDB | BLOSUM matrix | ANN | AUC, Sensitivity | None | |
| ACME [3] | IEDB, IPD-IMGT database | BLOSUM matrix | CNN | PCC, AUC | https://github.com/HYsxe/ACME | |
| NetMHCcons [42] | IEDB | Combination of NetMHC, NetMHCpan, PickPocket | None | PCC, t-test | www.cbs.dtu.dk/services/NetMHCcons | |
| NetMHCpan-4.0 [43] | IEDB | BLOSUM matrix | ANN | AUC | http://www.cbs.dtu.dk/services/NetMHCpan-4.0/ | |
| Lundegaard et al. [44] | IEDB | None | None | PCC, AUC | None | |
| Many-to-Many | MHCflurry [45] | IEDB, Other | BLOSUM matrix | ANN | AUC, KRCC, PCC, PPV | https://github.com/openvax/mhcflurry |
| CNN-NF [46] | IEDB, DB2013 | Sequence, Hydropathy, Polarity, Length | CNN | F1, AUC | https://github.com/zty2009/MHC-I/tree/master. |
| Training strategy | Models | Dataset | Feature representationa | Prediction modelb | Evaluation criteriac | Code or Webserver |
|---|---|---|---|---|---|---|
| One-to-One | SYFPEITHI [14] | EMBL, RDBMS | constructing motif pattern | Heuristic score | None | http://www.syfpeithi.de/ |
| ProPred1 [23] | MHCBN | QM | Heuristic score | Sensitivity, Specificity, Accuracy, MCC | http://www.imtech.res.in/raghava/propred1 | |
| SVMHC [24] | MHCPEP, SYFPEITHI, ENSEMBL | sparse encoding | SVM | MCC, Specificity, Sensitivity | http://www.sbc.su.se/svmhc/ | |
| SMMPMBEC [25] | Laboratories | PMBEC | SMM | AUC | http://www.mhc-pathway.net/smmpmbec. | |
| ConvMHC [26] | IEDB, Sette and Buus laboratories | physicochemical scores | CNN | F1, Precision, Sensitivity | http://jumong.kaist.ac.kr:8080/convmhc | |
| PickPocket [27] | IEDB | PSSM | Heuristic score | AUC, PCC | None | |
| Rasmussen et al. [28] | IEDB | BLOSUM matrix | ANN | AUC, PCC | http://www.cbs.dtu.dk/services/NetMHCstabpan | |
| Bhasin et al. [29] | MHCBN, SWISS-PROT | QM, BIMAS | ANN | Sensitivity, Specificity, Accuracy, Precision | https://webs.iiitd.edu.in/raghava/nhlapred/neural.html | |
| RANKPEP [30] | MHCPEP, SYFPEITHI | PSSM | Heuristic score | www.mifoundation.org/Tools/rankpep.html | ||
| ForestMHC [31] | PRIDE, SysteMHC, Other | BLOSUM matrix, one-hot, physicochemical | RF | AUPR, Prec1% | None | |
| Javadi et al. [32] | IEDB | physicochemical, composition | RF | AUC, Accuracy, Sensitivity, PPV, Specificity, NPV | None | |
| Wilson et al. [5] | IEDB, MassIVE | biochemical | RF | AUC, Accuracy, Sensitivity, Specificity | None | |
| Mattsson et al. [33] | IEDB, SYSPETHI | BLOSUM matrix | ANN | PCC | None | |
| One-to-Many | PSSMHCpan [34] | MHCBN, IEDB, SYFPEITHI, Laboratories | BLOSUM matrix, PSSM | Heuristic score | ACC, AUC | https://github.com/BGI2016/PSSMHCpan |
| NetMHC-3.0 [35] | IEDB, SYFPEITHI | PSSMs | ANN | PCC, AUC | http://www.cbs.dtu.dk/services/NetMHC-3.0 | |
| NetMHC-4.0 [36] | IEDB, SYFPEITHI | BLOSUM matrix, sparse encoding | ANN | PCC, AUC | http://www.cbs.dtu.dk/services/NetMHC-4.0 | |
| NIELSEN et al. [37] | SYFPEITHI | sparse encoding, BLOSUM matrix | ANN | PCC, AUC, Sensitivity/PPV | None | |
| NetMHCpan [38] | IEDB | sparse encoding, BLOSUM matrix | ANN | Specificity, Sensitivity, PCC | http://www.cbs.dtu.dk/services/NetMHCpan/ | |
| NetMHCpan-3.0 [39] | IEDB, SYFPEITHI | BLOSUM matrix | ANN-based ML | PCC, AUC | www.cbs.dtu.dk/services/NetMHCpan-3.0 | |
| HLA-CNN [40] | MHCBN, IEDB, AntiJen, SYFPEITHI | HLA-Vec | CNN | SRCC, AUC | https://github.com/uci-cbcl/HLA-bind. | |
| NetH2pan [41] | IEDB | BLOSUM matrix | ANN | AUC, Sensitivity | None | |
| ACME [3] | IEDB, IPD-IMGT database | BLOSUM matrix | CNN | PCC, AUC | https://github.com/HYsxe/ACME | |
| NetMHCcons [42] | IEDB | Combination of NetMHC, NetMHCpan, PickPocket | None | PCC, t-test | www.cbs.dtu.dk/services/NetMHCcons | |
| NetMHCpan-4.0 [43] | IEDB | BLOSUM matrix | ANN | AUC | http://www.cbs.dtu.dk/services/NetMHCpan-4.0/ | |
| Lundegaard et al. [44] | IEDB | None | None | PCC, AUC | None | |
| Many-to-Many | MHCflurry [45] | IEDB, Other | BLOSUM matrix | ANN | AUC, KRCC, PCC, PPV | https://github.com/openvax/mhcflurry |
| CNN-NF [46] | IEDB, DB2013 | Sequence, Hydropathy, Polarity, Length | CNN | F1, AUC | https://github.com/zty2009/MHC-I/tree/master. |
aPSSM, Position-Specific Scoring Matrix. BLOSUM, BLOcks SUbstitution Matrix. QM, Quantitative Matrix.
bANN, Artificial Neural Network. CNN, Convolutional Neural Networks . ML, Machine Learning. HMM, Hidden Markov Models. SVM, Support Vector Machine. RF, Random Forest.
cACC, Accuracy. AUC,Area Under the receiver-operating-characteristic Curve. AUPR, Area Under the Precision-Recall curve. MCC, Matthews Correlation Coefficient. PCC, Pearson Correlation Coefficient. SRCC, Spearman's Rank Correlation Coefficient. Prec1%, Precision in the top 1% of predictions. PPV, Positive Predictive Value. NPV, Negative Predictive Value.
A summary of 27 MHC prediction tools
| Training strategy | Models | Dataset | Feature representationa | Prediction modelb | Evaluation criteriac | Code or Webserver |
|---|---|---|---|---|---|---|
| One-to-One | SYFPEITHI [14] | EMBL, RDBMS | constructing motif pattern | Heuristic score | None | http://www.syfpeithi.de/ |
| ProPred1 [23] | MHCBN | QM | Heuristic score | Sensitivity, Specificity, Accuracy, MCC | http://www.imtech.res.in/raghava/propred1 | |
| SVMHC [24] | MHCPEP, SYFPEITHI, ENSEMBL | sparse encoding | SVM | MCC, Specificity, Sensitivity | http://www.sbc.su.se/svmhc/ | |
| SMMPMBEC [25] | Laboratories | PMBEC | SMM | AUC | http://www.mhc-pathway.net/smmpmbec. | |
| ConvMHC [26] | IEDB, Sette and Buus laboratories | physicochemical scores | CNN | F1, Precision, Sensitivity | http://jumong.kaist.ac.kr:8080/convmhc | |
| PickPocket [27] | IEDB | PSSM | Heuristic score | AUC, PCC | None | |
| Rasmussen et al. [28] | IEDB | BLOSUM matrix | ANN | AUC, PCC | http://www.cbs.dtu.dk/services/NetMHCstabpan | |
| Bhasin et al. [29] | MHCBN, SWISS-PROT | QM, BIMAS | ANN | Sensitivity, Specificity, Accuracy, Precision | https://webs.iiitd.edu.in/raghava/nhlapred/neural.html | |
| RANKPEP [30] | MHCPEP, SYFPEITHI | PSSM | Heuristic score | www.mifoundation.org/Tools/rankpep.html | ||
| ForestMHC [31] | PRIDE, SysteMHC, Other | BLOSUM matrix, one-hot, physicochemical | RF | AUPR, Prec1% | None | |
| Javadi et al. [32] | IEDB | physicochemical, composition | RF | AUC, Accuracy, Sensitivity, PPV, Specificity, NPV | None | |
| Wilson et al. [5] | IEDB, MassIVE | biochemical | RF | AUC, Accuracy, Sensitivity, Specificity | None | |
| Mattsson et al. [33] | IEDB, SYSPETHI | BLOSUM matrix | ANN | PCC | None | |
| One-to-Many | PSSMHCpan [34] | MHCBN, IEDB, SYFPEITHI, Laboratories | BLOSUM matrix, PSSM | Heuristic score | ACC, AUC | https://github.com/BGI2016/PSSMHCpan |
| NetMHC-3.0 [35] | IEDB, SYFPEITHI | PSSMs | ANN | PCC, AUC | http://www.cbs.dtu.dk/services/NetMHC-3.0 | |
| NetMHC-4.0 [36] | IEDB, SYFPEITHI | BLOSUM matrix, sparse encoding | ANN | PCC, AUC | http://www.cbs.dtu.dk/services/NetMHC-4.0 | |
| NIELSEN et al. [37] | SYFPEITHI | sparse encoding, BLOSUM matrix | ANN | PCC, AUC, Sensitivity/PPV | None | |
| NetMHCpan [38] | IEDB | sparse encoding, BLOSUM matrix | ANN | Specificity, Sensitivity, PCC | http://www.cbs.dtu.dk/services/NetMHCpan/ | |
| NetMHCpan-3.0 [39] | IEDB, SYFPEITHI | BLOSUM matrix | ANN-based ML | PCC, AUC | www.cbs.dtu.dk/services/NetMHCpan-3.0 | |
| HLA-CNN [40] | MHCBN, IEDB, AntiJen, SYFPEITHI | HLA-Vec | CNN | SRCC, AUC | https://github.com/uci-cbcl/HLA-bind. | |
| NetH2pan [41] | IEDB | BLOSUM matrix | ANN | AUC, Sensitivity | None | |
| ACME [3] | IEDB, IPD-IMGT database | BLOSUM matrix | CNN | PCC, AUC | https://github.com/HYsxe/ACME | |
| NetMHCcons [42] | IEDB | Combination of NetMHC, NetMHCpan, PickPocket | None | PCC, t-test | www.cbs.dtu.dk/services/NetMHCcons | |
| NetMHCpan-4.0 [43] | IEDB | BLOSUM matrix | ANN | AUC | http://www.cbs.dtu.dk/services/NetMHCpan-4.0/ | |
| Lundegaard et al. [44] | IEDB | None | None | PCC, AUC | None | |
| Many-to-Many | MHCflurry [45] | IEDB, Other | BLOSUM matrix | ANN | AUC, KRCC, PCC, PPV | https://github.com/openvax/mhcflurry |
| CNN-NF [46] | IEDB, DB2013 | Sequence, Hydropathy, Polarity, Length | CNN | F1, AUC | https://github.com/zty2009/MHC-I/tree/master. |
| Training strategy | Models | Dataset | Feature representationa | Prediction modelb | Evaluation criteriac | Code or Webserver |
|---|---|---|---|---|---|---|
| One-to-One | SYFPEITHI [14] | EMBL, RDBMS | constructing motif pattern | Heuristic score | None | http://www.syfpeithi.de/ |
| ProPred1 [23] | MHCBN | QM | Heuristic score | Sensitivity, Specificity, Accuracy, MCC | http://www.imtech.res.in/raghava/propred1 | |
| SVMHC [24] | MHCPEP, SYFPEITHI, ENSEMBL | sparse encoding | SVM | MCC, Specificity, Sensitivity | http://www.sbc.su.se/svmhc/ | |
| SMMPMBEC [25] | Laboratories | PMBEC | SMM | AUC | http://www.mhc-pathway.net/smmpmbec. | |
| ConvMHC [26] | IEDB, Sette and Buus laboratories | physicochemical scores | CNN | F1, Precision, Sensitivity | http://jumong.kaist.ac.kr:8080/convmhc | |
| PickPocket [27] | IEDB | PSSM | Heuristic score | AUC, PCC | None | |
| Rasmussen et al. [28] | IEDB | BLOSUM matrix | ANN | AUC, PCC | http://www.cbs.dtu.dk/services/NetMHCstabpan | |
| Bhasin et al. [29] | MHCBN, SWISS-PROT | QM, BIMAS | ANN | Sensitivity, Specificity, Accuracy, Precision | https://webs.iiitd.edu.in/raghava/nhlapred/neural.html | |
| RANKPEP [30] | MHCPEP, SYFPEITHI | PSSM | Heuristic score | www.mifoundation.org/Tools/rankpep.html | ||
| ForestMHC [31] | PRIDE, SysteMHC, Other | BLOSUM matrix, one-hot, physicochemical | RF | AUPR, Prec1% | None | |
| Javadi et al. [32] | IEDB | physicochemical, composition | RF | AUC, Accuracy, Sensitivity, PPV, Specificity, NPV | None | |
| Wilson et al. [5] | IEDB, MassIVE | biochemical | RF | AUC, Accuracy, Sensitivity, Specificity | None | |
| Mattsson et al. [33] | IEDB, SYSPETHI | BLOSUM matrix | ANN | PCC | None | |
| One-to-Many | PSSMHCpan [34] | MHCBN, IEDB, SYFPEITHI, Laboratories | BLOSUM matrix, PSSM | Heuristic score | ACC, AUC | https://github.com/BGI2016/PSSMHCpan |
| NetMHC-3.0 [35] | IEDB, SYFPEITHI | PSSMs | ANN | PCC, AUC | http://www.cbs.dtu.dk/services/NetMHC-3.0 | |
| NetMHC-4.0 [36] | IEDB, SYFPEITHI | BLOSUM matrix, sparse encoding | ANN | PCC, AUC | http://www.cbs.dtu.dk/services/NetMHC-4.0 | |
| NIELSEN et al. [37] | SYFPEITHI | sparse encoding, BLOSUM matrix | ANN | PCC, AUC, Sensitivity/PPV | None | |
| NetMHCpan [38] | IEDB | sparse encoding, BLOSUM matrix | ANN | Specificity, Sensitivity, PCC | http://www.cbs.dtu.dk/services/NetMHCpan/ | |
| NetMHCpan-3.0 [39] | IEDB, SYFPEITHI | BLOSUM matrix | ANN-based ML | PCC, AUC | www.cbs.dtu.dk/services/NetMHCpan-3.0 | |
| HLA-CNN [40] | MHCBN, IEDB, AntiJen, SYFPEITHI | HLA-Vec | CNN | SRCC, AUC | https://github.com/uci-cbcl/HLA-bind. | |
| NetH2pan [41] | IEDB | BLOSUM matrix | ANN | AUC, Sensitivity | None | |
| ACME [3] | IEDB, IPD-IMGT database | BLOSUM matrix | CNN | PCC, AUC | https://github.com/HYsxe/ACME | |
| NetMHCcons [42] | IEDB | Combination of NetMHC, NetMHCpan, PickPocket | None | PCC, t-test | www.cbs.dtu.dk/services/NetMHCcons | |
| NetMHCpan-4.0 [43] | IEDB | BLOSUM matrix | ANN | AUC | http://www.cbs.dtu.dk/services/NetMHCpan-4.0/ | |
| Lundegaard et al. [44] | IEDB | None | None | PCC, AUC | None | |
| Many-to-Many | MHCflurry [45] | IEDB, Other | BLOSUM matrix | ANN | AUC, KRCC, PCC, PPV | https://github.com/openvax/mhcflurry |
| CNN-NF [46] | IEDB, DB2013 | Sequence, Hydropathy, Polarity, Length | CNN | F1, AUC | https://github.com/zty2009/MHC-I/tree/master. |
aPSSM, Position-Specific Scoring Matrix. BLOSUM, BLOcks SUbstitution Matrix. QM, Quantitative Matrix.
bANN, Artificial Neural Network. CNN, Convolutional Neural Networks . ML, Machine Learning. HMM, Hidden Markov Models. SVM, Support Vector Machine. RF, Random Forest.
cACC, Accuracy. AUC,Area Under the receiver-operating-characteristic Curve. AUPR, Area Under the Precision-Recall curve. MCC, Matthews Correlation Coefficient. PCC, Pearson Correlation Coefficient. SRCC, Spearman's Rank Correlation Coefficient. Prec1%, Precision in the top 1% of predictions. PPV, Positive Predictive Value. NPV, Negative Predictive Value.
Strategies for existing MHC class I prediction methods
There are two major strategies for MHC binding prediction: machine learning and calculation score. Machine learning-based methods usually follow the following three steps: 1) feature representation; 2) training; and 3) evaluation. Calculation score based methods replace the training step with a scoring model. In this review, we discuss 27 MHC class I binding prediction methods. These methods’ classifications are available in Table 2.
Feature representation
Feature representation is a technique to digitally code properties of an amino acid sequence as an MHC binding target. Over the last three decades, many feature representation methods have been developed for predicting MHC binders. Here we discuss nine feature representation methods. Foremost, BLOcks SUbstitution Matrix (BLOSUM) [28, 34, 36–39, 41, 43] and position-specific scoring matrix (PSSM) [30, 34, 35, 44, 47–49] have been popular choices. With |$L$| denoting the length of the peptide in question, BLOSUM is a |$20\ (\mathrm{amino}\ \mathrm{acid})\times L$| matrix that represents substitution frequency between each pair of amino acids. PSSM is a similar |$20\ (\mathrm{amino}\ \mathrm{acid})\times L$| matrix that captures evolutionary conservation within amino acid pairs. The runtime of PSSM is substantially longer than BLOSUM because the PSSM needs to be extracted by comparing a database with large protein sequence. Stabilized matrix method (SMM) has been used in PickPocket [27] to construct the PSSM with shortened runtime. PickPocket incorporates information of the MHC molecule per se into the representation of the binders. Another widely used sequence representation method is one-hot encoding [50], which transforms a peptide sequence into a |$20\ (\mathrm{amino}\ \mathrm{acid})\times L$| binary matrix with one row denoting each amino acid. Another common method is quantitative matrix (QM), which considers the contribution of each amino acid in a peptide sequence [29]. QM can be obtained from BIMAS [51] server by inputting amino acid sequences.
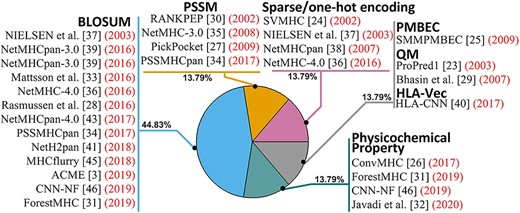
Usage frequency of feature representation methods to MHC prediction tools.
In addition to the four aforementioned conventional methods, other approaches have also been developed. For example, several studies proposed to use protein structure to model MHC class I binders. For example, Altuvia et al. [52] developed the first computational method based on protein structure to uncover MHC class I binders. Subsequently, Altuvia et al. [53] further improved the structure-based method to distinguish candidate peptides that bind to hydrophobic binding pockets of MHC molecules. Schueler-Furman et al. [54] extended the structure-based computational method to a wider range of MHC class I alleles. Other noticeable methods followed these pioneer works. Han et al. [26] merged information of the HLA molecule and the length of the target peptide into a |$34\times 9$| matrix, where 9 is the length of peptide and 34 is HLA-I contact residues proposed in NetMHCpan [38]. Kim et al. [25] developed the peptide:MHC binding energy covariance (PMBEC) matrix based on physicochemical properties (aromatic, hydrophobic and acidic etc.) to represent peptide sequence. Vang et al. [40] developed the HLA-Vec method to map amino acids to a 15-dimensional vector space to represent a peptide sequence. Bui et al. [55] constructed a |$20\ (\mathrm{amino}\ \mathrm{acid})\times L$| average relative binding coefficient matrix depending on geometric average binding affinity, or half maximal inhibitory concentration (IC50) of peptides.
Feature representation is a necessary component of MHC binding prediction tools. In one prediction tool, one or multiple feature presentation methods can be applied. In an evaluation study in 2003, Nielsen et al. [37] applied information from two feature presentation methods to predict MHC T-cell class I epitope and accordingly demonstrated that multiple sources of information can improve performance. Subsequent prediction tools generally exercised this concept. For example, DeepLigand [4] and NetMHCpan [38] used the combination of one-hot encoding and BLOSUM. ProPred1 [23] applied two types of QM from BIMAS. NetMHCpan 4.0 [43] combined BLOSUM and length of peptide flanking regions. Zhao et al. [46] combined BLOSUM and physicochemical properties. Nielsen et al. [37] integrated information from one-hot encoding, BLOSUM and Hidden Markov models (HMM). The incorporation of various feature representation methods into various binding prediction tools is illustrated in Figure 2.
Score based methods
Heuristic score-based methods [14, 23, 30, 34] came as classical approaches to predicting MHC binding. They calculate a quasi-probability affinity score for a peptide as a potential MHC binder, using three strategies: 1) sequence similarity [56, 57]; 2) motif incorporation [14, 58]; 3) matrix formulation [23, 34]. In the end, an affinity score or probability is used to decide binding potential with a threshold.
Sequence similarity based methods summarize the amino acid composition of known MHC binding peptides into a sequence profile, and then compute a MHC binding probability index for a new peptide [57]. Similar to sequence similarity-based methods, motif-based methods first construct a score motif based on known MHC binding peptides, then a new peptide is compared to the motif to obtain binding affinity score. The score motif generally assumes integer values, with a bigger number for a higher occurrence frequency of the amino acid. To construct motifs from known sequences, existing methods all require the peptides to have an equal length. For example, SYFPEITHI [14] constructs a |$20\ (\mathrm{amino}\ \mathrm{acid})\times 9$| motif pattern, by combining the information from MHC ligands, where 9 is the fixed peptide length. Here the motif score ranges from 1 to 10. Similarly, Sette et al. [58] constructed a |$20\ (\mathrm{amino}\ \mathrm{acid})\times 6$| substitution motif, where 6 represents the length of the core region of peptides. Here the motif score ranges from 1 to 3, with a smaller number indicating substitutions with drastic effects on |${IA}^d$| binding.
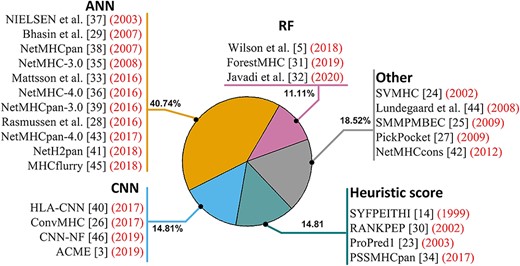
Usage frequency of machine learning models to MHC prediction tools.
Lastly, matrix-based methods differ from sequence similarity-based and motif-based methods by constructing a |$20\ (\mathrm{amino} \mathrm{acid})\times L$| matrix for a new peptide instead of known MHC binding peptides. For example, PSSMHCpan model [34] calculates a score to represent binding affinity based on PSSM. ProPred1 [23] uses QM to compute the score. Bui et al. [55] propose an average relative binding matrix to compute the score. All these methods proceed to convert the scores to quasi-IC50 values for making a binary decision with 500 as a typical threshold for IC50.
Traditional machine learning based methods
Traditional machine learning methods are frequently applied to develop MHC binding prediction tools. For example, SVMHC [24] encodes peptide sequence with one-hot encoding and uses support vector machine to predict MHC class I binding peptides. Liu et al. [59] applied physicochemical properties to a support vector machine regression model to construct the SVRMHC model for predicting binding affinity on mouse MHC I molecules. Kevin et al. [31], Javadi et al. [32] and Wilson et al. [5] used random forest (RF) model to predict immunogenic peptides, where a peptide sequence is encoded mainly through physicochemical properties. To improve the performance of MHC binding prediction, SMMPMBEC [25] applies a Bayesian framework to work with the peptide MHC binding energy covariance (PMBEC) similarity matrix.
Network-based machine learning has also been applied in MHC prediction. Luo et al. [60] constructed a network between HLA molecules and peptides, and thereby devised Nebula, a neighbor-edges-based and unbiased leverage algorithm, to discover new HLA–peptide binding. Later, Luo et al. [61] improved Nebula model to a derivative named sNebula.
Yet another category of machine learning methods frequently applied in MHC binding prediction is Artificial Neural Networks (ANN). In 2008, Lin et al. [62] demonstrated that ANN has better performance than score-based methods by comparatively evaluating 30 methods. Since then, multiple ANN-based tools have been developed. For example, NetMHC-3.0 [35] establishes a high-performance ANN web server for predicting peptide binders. NetMHCpan tools [38, 39, 43, 63] and MHCflurry [45] also used ANN to predict MHC binders, achieving excellent performance on multiple species. To prevent overfitting, NetMHCpan explored multiple ANN models with different number of neuron nodes and culminated on the optimal model with the highest area under the curve (AUC) or Pearson correlation coefficient (PCC) in 5-fold cross-validation. To improve the predicting performance of ANN, Nielsen et al. [37] and Zhang et al. [64] combined HMM and ANN to distinguish the MHC binding peptides. The dissection of MHC prediction tools by traditional machine learning models is illustrated in Figure 3.
Deep learning methods
Convolutional neural networks (CNN) [26, 40, 46] and Deep Residual Network [4] are two widely used deep learning models for predicting MHC binders. For example, HLA-CNN [40] and ConvMHC [26] use a CNN architecture and fully connected layers to predict MHC class I binders. DeepSeqPan [65] is a CNN based multi-task tool to calculate an IC50 affinity value and a binding probability simultaneously, which derive its feature information from the paired peptide and HLA molecule. Another CNN-based tool CNN-NF [46] is composed of two activation layers, two pool layers, one flatten layer and one full connection layer. DeepLigand [4] applies Deep Residual Network to construct an affinity prediction module and a peptide embedding module and then uses a fully connected layer to connect the two sub-modules for prediction.
Machine learning training strategy
Based on the sequence length of MHC binders, training strategies of machine learning methods can be classified into three categories of distinct training strategies: 1) One-to-One [14, 23, 24, 28, 62, 66, 67], 2) One-to-Many [34, 38, 40–42, 49] and 3) Many-to-Many [45, 46] (Figure 4).
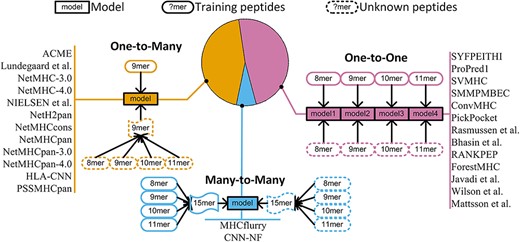
Categorization of MHC prediction tools into three training strategies.
The One-to-One strategy limits the model to training peptides with the sample length. Thus, models of this type can only predict peptides of the same length as the model. Multiple models are necessary to predict peptides of various lengths. The most popular length choice for the One-to-One strategy is 9-mer [68], because in most cases binding affinity is measured for 9-mer peptides. The One-to-Many strategy was developed to overcome the challenge of length dependency. In 2008, Lundegaard et al. [44] proposed a strategy to encode 8-mer and10-mer peptides in addition to 9-mer peptides by considering insertion and deletion of amino acids. Subsequent tools [3, 34–36, 38, 39] extended the same idea to 11-mer peptides. Insertion and deletion of amino acids in One-to-Many and Many-to-Many strategies are illustrated in Figure 5.
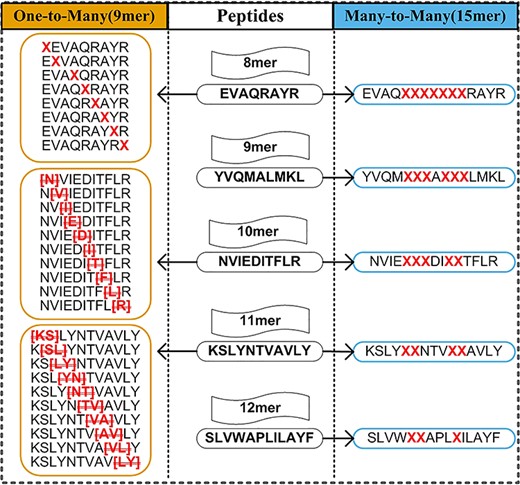
Changing length of peptides in One-to-Many and Many-to-Many strategies. For One-to-Many, 8-mer, 10-mer and 11-mer peptides are changed to 9-mer peptide by inserting ‘X’ character and deleting amino acid. For Many-to-Many, 8–12-mer peptides are changed to 15-mer peptide by inserting ‘X’ character.
Even though the One-to-Many strategy can be used to predict MHC binders with different amino acids, the models are trained on 9-mer peptides only. In addition, One-to-Many is limited to a maximum length of 11. The Many-to-Many strategy is a relatively novel approach. The MHCflurry [45] model proposed by O'Donnell et al. in 2018 encodes peptides of up to 15 amino acids as 15-mer sequences by inserting ‘X’ character, and uses 15-mer BLOSUM matrix to train the model. When inserting the ‘X’ characters, MHCflurry requires that no ‘X’ can be inserted into the first and last four amino acids of the peptide (Figure 5). Zhao et al. [46] applied the same principle as MHCflurry but trained the model with the CNN-NF algorithm. The Many-to-Many strategy has strength in that it allows the training of a single model for peptides of various lengths. Although these strategies to accommodate variable lengths have achieved good performance to predict MHC binders, the insertion or deletion of amino acids in peptide sequence can potentially alter the primary structure and thus lead to loss of information. It is worthwhile to train a length-independent model, which preserves peptide sequence structure. With this objective in mind, we developed BVLSTM-MHC, a bilateral and variable recurrent neural network-based method to overcome this limitation.
Evaluation parameters
BVLSTM for MHC binder prediction
Datasets
Peptide–MHC binding relations from IEDB were extracted to train BVLSTM-MHC. Benchmark data, including 117 alleles of four species (Mouse, Human, Macaque and Chimpanzee), were also downloaded from IEDB. The sequence length of the IEDB entries ranges from 8 to 18 amino acids. An independent validation dataset, consisting of 42 MHC binders and 179 non-binders, was extracted from MHCBN [11]. Organization of positive examples (binders) and negative examples (binders) for MHC allele classes of four species is shown in Table 3 and Supplementary Figure 1, where species-omitted notations HLA-A, HLA-B and HLA-C all relate to human.
Details of training data from IEDB and validation data from MHCBN
| dataset | Species | MHC Allele class | Number of Alleles | 9-mer binders | 9-mer non-binder | non-9-mer binders | non-9-mer non-binder |
|---|---|---|---|---|---|---|---|
| IEDB dataset | Mouse | H-2 | 6 | 1375 | 3165 | 1343 | 3899 |
| Human | HLA-A | 37 | 19,716 | 52,835 | 8438 | 16,640 | |
| HLA-B | 39 | 9367 | 36,256 | 2317 | 7510 | ||
| HLA-C | 9 | 871 | 1163 | 8 | 162 | ||
| Macaque | Mamu-A Mamu-B | 18 | 2716 | 3568 | 2256 | 5486 | |
| Chimpanzee | Patr-A Patr-B | 8 | 514 | 1440 | 544 | 1204 | |
| Total | 117 | 34,559 | 98,427 | 14,906 | 34,901 | ||
| MHCBN dataset | Human | HLA-A | 10 | 13 | 106 | 10 | 30 |
| HLA-B | 5 | 8 | 22 | 11 | 21 | ||
| Total | 15 | 21 | 128 | 21 | 51 |
| dataset | Species | MHC Allele class | Number of Alleles | 9-mer binders | 9-mer non-binder | non-9-mer binders | non-9-mer non-binder |
|---|---|---|---|---|---|---|---|
| IEDB dataset | Mouse | H-2 | 6 | 1375 | 3165 | 1343 | 3899 |
| Human | HLA-A | 37 | 19,716 | 52,835 | 8438 | 16,640 | |
| HLA-B | 39 | 9367 | 36,256 | 2317 | 7510 | ||
| HLA-C | 9 | 871 | 1163 | 8 | 162 | ||
| Macaque | Mamu-A Mamu-B | 18 | 2716 | 3568 | 2256 | 5486 | |
| Chimpanzee | Patr-A Patr-B | 8 | 514 | 1440 | 544 | 1204 | |
| Total | 117 | 34,559 | 98,427 | 14,906 | 34,901 | ||
| MHCBN dataset | Human | HLA-A | 10 | 13 | 106 | 10 | 30 |
| HLA-B | 5 | 8 | 22 | 11 | 21 | ||
| Total | 15 | 21 | 128 | 21 | 51 |
Details of training data from IEDB and validation data from MHCBN
| dataset | Species | MHC Allele class | Number of Alleles | 9-mer binders | 9-mer non-binder | non-9-mer binders | non-9-mer non-binder |
|---|---|---|---|---|---|---|---|
| IEDB dataset | Mouse | H-2 | 6 | 1375 | 3165 | 1343 | 3899 |
| Human | HLA-A | 37 | 19,716 | 52,835 | 8438 | 16,640 | |
| HLA-B | 39 | 9367 | 36,256 | 2317 | 7510 | ||
| HLA-C | 9 | 871 | 1163 | 8 | 162 | ||
| Macaque | Mamu-A Mamu-B | 18 | 2716 | 3568 | 2256 | 5486 | |
| Chimpanzee | Patr-A Patr-B | 8 | 514 | 1440 | 544 | 1204 | |
| Total | 117 | 34,559 | 98,427 | 14,906 | 34,901 | ||
| MHCBN dataset | Human | HLA-A | 10 | 13 | 106 | 10 | 30 |
| HLA-B | 5 | 8 | 22 | 11 | 21 | ||
| Total | 15 | 21 | 128 | 21 | 51 |
| dataset | Species | MHC Allele class | Number of Alleles | 9-mer binders | 9-mer non-binder | non-9-mer binders | non-9-mer non-binder |
|---|---|---|---|---|---|---|---|
| IEDB dataset | Mouse | H-2 | 6 | 1375 | 3165 | 1343 | 3899 |
| Human | HLA-A | 37 | 19,716 | 52,835 | 8438 | 16,640 | |
| HLA-B | 39 | 9367 | 36,256 | 2317 | 7510 | ||
| HLA-C | 9 | 871 | 1163 | 8 | 162 | ||
| Macaque | Mamu-A Mamu-B | 18 | 2716 | 3568 | 2256 | 5486 | |
| Chimpanzee | Patr-A Patr-B | 8 | 514 | 1440 | 544 | 1204 | |
| Total | 117 | 34,559 | 98,427 | 14,906 | 34,901 | ||
| MHCBN dataset | Human | HLA-A | 10 | 13 | 106 | 10 | 30 |
| HLA-B | 5 | 8 | 22 | 11 | 21 | ||
| Total | 15 | 21 | 128 | 21 | 51 |
BVLSTM-MHC model
Here, we develop a recurrent neural network framework of strong scalability, BVLSTM-MHC, to predict MIC class I binders (Figure 6). MHC binders are first transformed into a |$20\ (\mathrm{amino}\ \mathrm{acid})\times L$| matrix with one-hot encoding, where L is the number of amino acids of MHC binder. Then, one-hot encoding matrix and BLOSUM are combined to a |$20\times L$| matrix to represent the peptide sequence. BLOSUM is used to initialize 20 convolution kernels. This process is shown in the section of the feature extraction layer in Figure 6. Then, the merged matrix is put into BVLSTM Encode Model, which is composed of two sets of LSTM. In the BVLSTM Encode Model, LSTM block dynamically changes with the sequence length. One set of LSTM processes the input matrix from left to right and the other set of LSTM processes the input matrix from right to left. This processing mechanism makes handling peptides of different lengths in one same model possible. Then, two fully-connected layers with 64 and 1 neurons, respectively, are used to handle the output vector of BVLSTM model. Finally, the Sigmoid function is used to normalize the output value to the predicted probability. All allele models shared these two fully-connected layers. In the training process, a dropout layer is applied to avoid over-fitting. The mean squared error is applied as the loss function. In the final models, the learning rate and the parameter of the dropout layer were set at |${10}^{-4}$| and 0.8, respectively, while a survey of a wide range of learning rates and dropout rates layer were shown in Supplementary Figure 2.

Schema of BVLSTM-MHC. Two major processes are detailed, namely feature representation and BVLSTM Encode model.
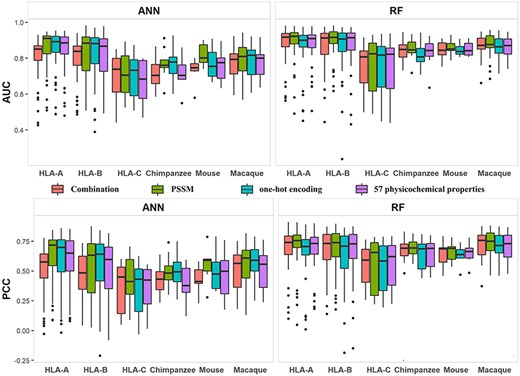
Distribution of AUCs and PCCs returned by four feature representation methods. The four methods include PSSM, one-hot encoding, 57 physicochemical properties and combination of the former three (combination).
Evaluation of four feature representation methods
Four feature representation approaches, one-hot encoding, PSSM, 57 physicochemical properties [69], and the combination of the three were compared with the IEDB dataset. PSSM was extracted by NCBI blast-2.2.29 on UniProt [70] database. ANN and RF were performed to analyze the four approaches on 9-mer peptides using five-fold cross-validation. Hidden neurons in ANN were set to 15 and the number of tree in RF was set to 500. PCC and AUC were used to assess the performance and results were shown in Figure 7. Data of 117 alleles were classified into six categories: HLA-A, HLA-B, HLA-C, Chimpanzee, Mouse and Macaque, where the first three categories belonged to Human. For ANN, the feature representation method PSSM achieved the highest medians for AUC and PCC in three categories. The combination of three individual methods achieved the best medians for AUC and PCC on the category of HLA-C, and One-hot encoding achieved the best medians of AUC and PCC for the categories of HLA-B and Chimpanzee. For RF, PSSM achieved the highest medians of AUC and PCC in five categories. The combination of three individual methods achieved the best performance of the median of AUC and PCC on the category of Chimpanzee and Mouse, respectively. Overall, PSSM had the best performance. Our analysis results also demonstrated that RF had higher AUCs and PCCs than ANN (Figure 7). This may be due to the fact that we used a fixed number of hidden neurons, as opposed to a variable number of hidden neurons.
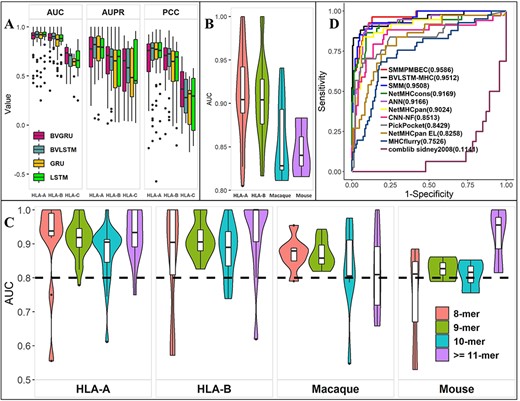
Prediction performance of the BVLSTM-MHC model in cross-validation. (A) Distribution of various performance metrics (AUC, AUPR and PCC) for various recurrent neural network models (BVGRU, BVLSTM, GRU and LSTM), stratified by MHC allele classes (HLA-A, HLA-B and HLA-C). (B) Distribution of AUC of the BVLSTM-MHC model on the test dataset. Results for chimpanzee and HLA-C were not plotted due to limited data points. (C) Distribution of AUC of the BVLSTM-MHC model on the test dataset, stratified by peptide length. Results for chimpanzee and HLA-C were not plotted due to limited data points. (D) Receiver-operating-characteristic curves of BVLSTM together with another 10 competitors. AUC values were annotated to each method.
Evaluation of four recurrent neural network models
We compared the performance of BVLSTM with three RNN models: bilateral variable gated recurrent units (BVGRU), LSTM and GRU. BVLSTM and BVGRU are bilateral variable-length models. LSTM and GRU are single-direction, fixed-length models. Four models were trained on 85 human alleles with 9-mer peptides. Bilateral Variable models performed better than LSTM and GRU models (Figure 8A). BVLSTM obtained the highest medians for AUC and PCC on HLA-A and HLA-B categories and the highest medians for AUPR on the HLA-A category. These results indicate that BVLSTM is a potentially optimal approach for predicting MHC binder peptides.
Cross-validation performance of BVLSTM-MHC
The IEDB dataset was split into 10 portions: seven were used for training, one was used as validation to select the optimal model, and the rest two were used for final testing. After five-fold cross-validation and validation, the final test results were as follows. Of the 117 alleles, 77 had AUC higher than 0.8 (62 human, 10 macaque, 3 mouse and 2 chimpanzee) (Figure 8B and Supplementary Table 1). For human HLA-A and HLA-B datasets, the medians of AUC were greater than 0.9 on both test datasets. The medians of AUC were above 0.8 for mouse, chimpanzee and HLA-C. AUCs of peptides with different lengths (8–18mers) were calculated to evaluate the variable-length performance of BVLSTM-MHC (Figure 8C and Supplementary Table 1). For human HLA-A and HLA-B, the median AUCs were greater than 0.9 regardless of peptide length. For HLA-C, mouse and chimpanzee, the median AUC of variable length peptides were closer to 0.8.
Performance of BVLSTM-MHC along with ten existent MHC class I predictors on independent MHCBN dataset
| Methods | ACC | AUC | F1 | MCC | Specificity | Sensitivity | Precision | AUPR | # Postive examples | # Negative examples |
|---|---|---|---|---|---|---|---|---|---|---|
| ANN [44] | 0.8827 | 0.9166 | 0.6866 | 0.6205 | 0.9507 | 0.6216 | 0.7667 | 0.7726 | 37 | 142 |
| comblibsidney 2008 [71] | 0.7419 | 0.1141 | — | 0.0000 | 1.0000 | 0.0000 | 0.0000 | 0.1591 | 16 | 46 |
| NetMHCcons [42] | 0.8883 | 0.9169 | 0.7222 | 0.6528 | 0.9366 | 0.7027 | 0.7429 | 0.8457 | 37 | 142 |
| NetMHCpan [63] | 0.8547 | 0.9024 | 0.5938 | 0.5173 | 0.9437 | 0.5135 | 0.7037 | 0.7471 | 37 | 142 |
| NetMHCpan EL [43] | 0.8156 | 0.8258 | 0.5075 | 0.3989 | 0.9085 | 0.4595 | 0.5667 | 0.5977 | 37 | 142 |
| PickPocket [27] | 0.8715 | 0.8429 | 0.6849 | 0.6043 | 0.9225 | 0.6757 | 0.6944 | 0.6378 | 37 | 142 |
| SMM [72] | 0.9268 | 0.9508 | 0.7692 | 0.7266 | 0.9635 | 0.7407 | 0.8000 | 0.8548 | 27 | 137 |
| SMMPMBEC [25] | 0.9146 | 0.9586 | 0.7308 | 0.6808 | 0.9562 | 0.7037 | 0.7600 | 0.8581 | 27 | 137 |
| BVLSTM-MHC | 0.9548 | 0.9512 | 0.8750 | 0.8490 | 0.9832 | 0.8333 | 0.9211 | 0.9112 | 42 | 179 |
| CNN-NF [46] | 0.8606 | 0.8513 | 0.6234 | 0.5449 | 0.8908 | 0.7059 | 0.5581 | 0.6383 | 34 | 174 |
| MHCflurry [45] | 0.7834 | 0.7526 | 0.4946 | 0.3633 | 0.8352 | 0.5610 | 0.4423 | 0.4569 | 41 | 176 |
| Methods | ACC | AUC | F1 | MCC | Specificity | Sensitivity | Precision | AUPR | # Postive examples | # Negative examples |
|---|---|---|---|---|---|---|---|---|---|---|
| ANN [44] | 0.8827 | 0.9166 | 0.6866 | 0.6205 | 0.9507 | 0.6216 | 0.7667 | 0.7726 | 37 | 142 |
| comblibsidney 2008 [71] | 0.7419 | 0.1141 | — | 0.0000 | 1.0000 | 0.0000 | 0.0000 | 0.1591 | 16 | 46 |
| NetMHCcons [42] | 0.8883 | 0.9169 | 0.7222 | 0.6528 | 0.9366 | 0.7027 | 0.7429 | 0.8457 | 37 | 142 |
| NetMHCpan [63] | 0.8547 | 0.9024 | 0.5938 | 0.5173 | 0.9437 | 0.5135 | 0.7037 | 0.7471 | 37 | 142 |
| NetMHCpan EL [43] | 0.8156 | 0.8258 | 0.5075 | 0.3989 | 0.9085 | 0.4595 | 0.5667 | 0.5977 | 37 | 142 |
| PickPocket [27] | 0.8715 | 0.8429 | 0.6849 | 0.6043 | 0.9225 | 0.6757 | 0.6944 | 0.6378 | 37 | 142 |
| SMM [72] | 0.9268 | 0.9508 | 0.7692 | 0.7266 | 0.9635 | 0.7407 | 0.8000 | 0.8548 | 27 | 137 |
| SMMPMBEC [25] | 0.9146 | 0.9586 | 0.7308 | 0.6808 | 0.9562 | 0.7037 | 0.7600 | 0.8581 | 27 | 137 |
| BVLSTM-MHC | 0.9548 | 0.9512 | 0.8750 | 0.8490 | 0.9832 | 0.8333 | 0.9211 | 0.9112 | 42 | 179 |
| CNN-NF [46] | 0.8606 | 0.8513 | 0.6234 | 0.5449 | 0.8908 | 0.7059 | 0.5581 | 0.6383 | 34 | 174 |
| MHCflurry [45] | 0.7834 | 0.7526 | 0.4946 | 0.3633 | 0.8352 | 0.5610 | 0.4423 | 0.4569 | 41 | 176 |
Performance of BVLSTM-MHC along with ten existent MHC class I predictors on independent MHCBN dataset
| Methods | ACC | AUC | F1 | MCC | Specificity | Sensitivity | Precision | AUPR | # Postive examples | # Negative examples |
|---|---|---|---|---|---|---|---|---|---|---|
| ANN [44] | 0.8827 | 0.9166 | 0.6866 | 0.6205 | 0.9507 | 0.6216 | 0.7667 | 0.7726 | 37 | 142 |
| comblibsidney 2008 [71] | 0.7419 | 0.1141 | — | 0.0000 | 1.0000 | 0.0000 | 0.0000 | 0.1591 | 16 | 46 |
| NetMHCcons [42] | 0.8883 | 0.9169 | 0.7222 | 0.6528 | 0.9366 | 0.7027 | 0.7429 | 0.8457 | 37 | 142 |
| NetMHCpan [63] | 0.8547 | 0.9024 | 0.5938 | 0.5173 | 0.9437 | 0.5135 | 0.7037 | 0.7471 | 37 | 142 |
| NetMHCpan EL [43] | 0.8156 | 0.8258 | 0.5075 | 0.3989 | 0.9085 | 0.4595 | 0.5667 | 0.5977 | 37 | 142 |
| PickPocket [27] | 0.8715 | 0.8429 | 0.6849 | 0.6043 | 0.9225 | 0.6757 | 0.6944 | 0.6378 | 37 | 142 |
| SMM [72] | 0.9268 | 0.9508 | 0.7692 | 0.7266 | 0.9635 | 0.7407 | 0.8000 | 0.8548 | 27 | 137 |
| SMMPMBEC [25] | 0.9146 | 0.9586 | 0.7308 | 0.6808 | 0.9562 | 0.7037 | 0.7600 | 0.8581 | 27 | 137 |
| BVLSTM-MHC | 0.9548 | 0.9512 | 0.8750 | 0.8490 | 0.9832 | 0.8333 | 0.9211 | 0.9112 | 42 | 179 |
| CNN-NF [46] | 0.8606 | 0.8513 | 0.6234 | 0.5449 | 0.8908 | 0.7059 | 0.5581 | 0.6383 | 34 | 174 |
| MHCflurry [45] | 0.7834 | 0.7526 | 0.4946 | 0.3633 | 0.8352 | 0.5610 | 0.4423 | 0.4569 | 41 | 176 |
| Methods | ACC | AUC | F1 | MCC | Specificity | Sensitivity | Precision | AUPR | # Postive examples | # Negative examples |
|---|---|---|---|---|---|---|---|---|---|---|
| ANN [44] | 0.8827 | 0.9166 | 0.6866 | 0.6205 | 0.9507 | 0.6216 | 0.7667 | 0.7726 | 37 | 142 |
| comblibsidney 2008 [71] | 0.7419 | 0.1141 | — | 0.0000 | 1.0000 | 0.0000 | 0.0000 | 0.1591 | 16 | 46 |
| NetMHCcons [42] | 0.8883 | 0.9169 | 0.7222 | 0.6528 | 0.9366 | 0.7027 | 0.7429 | 0.8457 | 37 | 142 |
| NetMHCpan [63] | 0.8547 | 0.9024 | 0.5938 | 0.5173 | 0.9437 | 0.5135 | 0.7037 | 0.7471 | 37 | 142 |
| NetMHCpan EL [43] | 0.8156 | 0.8258 | 0.5075 | 0.3989 | 0.9085 | 0.4595 | 0.5667 | 0.5977 | 37 | 142 |
| PickPocket [27] | 0.8715 | 0.8429 | 0.6849 | 0.6043 | 0.9225 | 0.6757 | 0.6944 | 0.6378 | 37 | 142 |
| SMM [72] | 0.9268 | 0.9508 | 0.7692 | 0.7266 | 0.9635 | 0.7407 | 0.8000 | 0.8548 | 27 | 137 |
| SMMPMBEC [25] | 0.9146 | 0.9586 | 0.7308 | 0.6808 | 0.9562 | 0.7037 | 0.7600 | 0.8581 | 27 | 137 |
| BVLSTM-MHC | 0.9548 | 0.9512 | 0.8750 | 0.8490 | 0.9832 | 0.8333 | 0.9211 | 0.9112 | 42 | 179 |
| CNN-NF [46] | 0.8606 | 0.8513 | 0.6234 | 0.5449 | 0.8908 | 0.7059 | 0.5581 | 0.6383 | 34 | 174 |
| MHCflurry [45] | 0.7834 | 0.7526 | 0.4946 | 0.3633 | 0.8352 | 0.5610 | 0.4423 | 0.4569 | 41 | 176 |
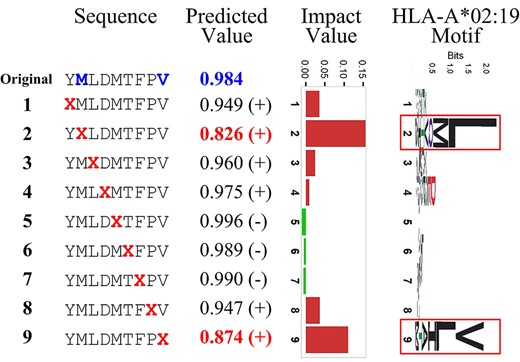
Binding prediction of BVLSTM-MHC is largely correlated with evolutionary conservation of amino acid positions. Using a 9-mer peptide of YMLDMTFPV sequence and the motif of allele HLA-A*02:19 as an example, we partially interpret the rationale of BVLSTM-MHC predicting MHC binding. For each position, we replace the amino acid with an ‘X’ character designating an unknown amino acid, thus generating 9 modulated sequences (‘Sequence’ column). BVLSTM-MHC reported a new binding probability score for each modulated sequence (‘Predicted Value’ column). The difference of MHC binding probability score between the original case and the modulated case was obtained and regarded as the impact value of each individual amino acid position (‘Impact Value’ column). Of note, we indicated decreased binding probability with red bars and increased binding probability with green bars. The two amino acid positions that displayed the highest impact values (the second and ninth positions) were highlighted in red for their predicted values. Referring to the sequence logo of the binding motif (the rightmost column), it is evident that BVLSTM-MHC model predicts binding propensity largely based on the evolutionary conservation of amino acid positions.
Performance of BVLSTM-MHC in comparison with existent MHC class I prediction methods
BVLSTM-MHC was compared to 10 popular MHC class I predictors: ANN [44], comblibsidney2008 [71], NetMHCcons [42], NetMHCpan [63], NetMHCpan EL [43], PickPocket [27], SMM [72], SMMPMBEC [25], CNN-NF [46] and MHCflurry [45] (Figure 8D). MHCBN dataset was used as an independent evaluation. Eight evaluation criteria (AUC, ACC, F1, MCC, specificity, sensitivity, precision and AUPR) were computed. The overall results can be viewed in Table 4 and the length specific results can be viewed in Supplementary Table 2. SMMPMBEC and obtained the best AUC (0.96), BVLSTM-MHC was closely behind with an AUC of 0.95. BVLSTM-MHC achieved the best performance for the other six parameters.
Association between BVLSTM-MHC and positional conservation
Despite high performance, deep learning models are often criticized for the lack of interpretability as a black box. Multiple tools such as LIME [73], SHAP [74] and DeepLIFT [75] have been designed to help understand the underlying mechanism of deep learning models. Unfortunately, our BVLSTM-MHC models were developed using a TensorFlow class that is not supported by existing interpretation tools yet. Nevertheless, inspired by these tools, we developed a strategy to interpret BVLSTM-MHC rationale. Using an exemplar 9-mer peptide and the BVLSTM-MHC model for allele HLA-A*02:19 as an example, we interpreted the rationale of BVLSTM-MHC prediction (Figure 9). Given the sequence logo for the motif of allele HLA-A*02:19 (extracted by ggseqlogo R package [76]), it was revealed that the impact of each position captured by BVLSTM-MHC matches the position-wise conservation level. This suggests that BVLSTM-MHC model predicts binding propensity largely based on the evolutionary conservation of amino acid positions.
BVLSTM-MHC webserver
Given all the above parameters and component optimization, we developed an online server BVLSTM-MHC to perform MHC binder prediction. Because we reserved only MHC alleles showing an interim AUC of 0.8 or higher, the ultimate web server spans four species (Human, mouse, macaque and chimpanzee) and covers 77 MHC class I alleles in total. BVLSTM-MHC was developed with R, PHP and Python languages, and it is accessible at http://www.innovebioinfo.com/Proteomics/MHCBIB/MHCI.php.
Discussion
MHC binding prediction is a crucial step for identifying potential novel therapeutic strategies. Many tools have been developed for this purpose. We introduced BVLSTM-MHC, a variable-length BVLSTM RNN based method, to predict MHC class I binders. In comparison to the 10 existent MHC class I binding prediction tools, BVLSTM-MHC performed best in six of eight evaluation parameters on an independent dataset. Most MHC class I predictors limited the maximum length of the peptides to 11 or 12, whereas BVLSTM-MHC currently extends maximum peptide length to 30, and the built-in variable-length design spares users the trouble to switch models of different length parameter. BVLSTM-MHC performed the best overall in six of the eight performance parameters when compared to 10 existing MHC class I prediction tools. The reason for performance improvement is probably attributed to the enlarged sample size and the ability to handle variable length without modifying peptide sequences. For the One-to-One model, it is necessary to build a prediction model for each length, thus inevitably limiting the sample size of each length-specific model. For the One-to-Many model, 9-mer peptides are used to train a prediction model and peptides with non-9-mer can be predicted by inserting ‘X’ character or deleting amino acids in the peptide sequence. For the Many-to-Many model, peptides of different lengths are all padded up to a uniform 15-mer length by inserting ‘X’ characters. One-to-Many and Many-to-Many models potentially alter the primary structure thus leading to loss of information, because primary structure determines tertiary structure to a large extent. Overall, this work yielded a web server based on the BVLSTM-MHC models, which can predict MHC class I binders of 77 alleles from four species.
We comprehensively summarized 13 MHC databases and 27 prediction tools for MHC class I binding.
We developed a variable-length MHC class I binding prediction tool based on remaining original peptide sequence, BVLSTM-MHC.
BVLSTM-MHC performed best in six out of eight evaluated metrics when compared to the 10 mainstream MHC class I binding predictors.
We developed a web server based on BVLSTM-MHC to predict human, mouse, macaque and chimpanzee MHC Class I alleles.
Funding
The National Natural Science Foundation of China (NSFC 61772362, 61972280); the National Key R&D Program of China (2020YFA0908400); the National Cancer Institute, USA (Grants P30CA118100 and R01ES030993-01A1 to Y.G.).
Data Availability
Data are available at http://www.innovebioinfo.com/Proteomics/MHCBIB/MHCI.php.
Limin Jiang is a PhD candidate at Tianjin University. Her research interests include Bioinformatics and Machine Learning.
Hui Yu is a research fellow at the Department of Internal Medicine, University of New Mexico.
Jiawei Li is currently a master's degree candidate at Tianjin University. His research interests include Deep Learning and Machine Learning.
Jijun Tang is a professor at the University of South Carolina. His main research interests include Computational Biology and Algorithm.
Yan Guo is an associate professor in the Department of Internal Medicine, University of New Mexico. He is also the director of Bioinformatics Shared Resources of the University of New Mexico, Comprehensive Cancer Center.
Fei Guo is a professor at Central South University. Her research interests include Bioinformatics and Computational Biology.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}