




Abstract
Meiotic recombination is one of the most important driving forces of biological evolution, which is initiated by double-strand DNA breaks. Recombination has important roles in genome diversity and evolution. This review firstly provides a comprehensive survey of the 15 computational methods developed for identifying recombination hotspots in Saccharomyces cerevisiae. These computational methods were discussed and compared in terms of underlying algorithms, extracted features, predictive capability and practical utility. Subsequently, a more objective benchmark data set was constructed to develop a new predictor iRSpot-Pse6NC2.0 (http://lin-group.cn/server/iRSpot-Pse6NC2.0). To further demonstrate the generalization ability of these methods, we compared iRSpot-Pse6NC2.0 with existing methods on the chromosome XVI of S. cerevisiae. The results of the independent data set test demonstrated that the new predictor is superior to existing tools in the identification of recombination hotspots. The iRSpot-Pse6NC2.0 will become an important tool for identifying recombination hotspot.
Introduction
Meiotic recombination is one of the most important driving forces of biological evolution, which is initiated by double-strand DNA breaks (DSBs) [1, 2]. Genetic recombination describes the generation of new combinations of alleles that occurs at each generation in diploid organisms [3, 4]. Thus, recombination has important roles in genome diversity and evolution. For example, it can help homologous chromosomes pair and alter genome structure by disrupting linkage of sequence polymorphisms on the same DNA molecule [5–8].
Recombination does not occur randomly on chromosomes. Some genomic regions in which meiotic DSBs occur at relatively high frequencies are called recombination hotspots, while the other regions with a lower frequency of DSBs occurrence are referred to as recombination coldspots. Requirements to form DSBs include open chromatin structure, presence of certain histone modifications and binding of sequence-specific transcription factors at some loci [9, 10]. Formation of DNA DSBs also requires the covalent binding of topoisomerase-related Spo11 protein to small DNA fragments [11].
Investigations on recombination events and identification of hotspots are significant for understanding the mechanism of recombination initiation in Saccharomyces cerevisiae. Traditionally, the discovery of recombination hotspots mainly relies on experimental methods. Several studies have been carried out for the in-depth study of recombination hotspots. Gerton et al. [1] used DNA microarray as the single-gene resolution method to estimate the DSBs formation adjacent to each Open Reading Frame (ORF) for the S. cerevisiae. Pan et al. [11] uncovered a genome-wide DSBs map of unprecedented resolution and sensitivity by sequencing the small DNA fragments that bound to accumulation of Spo11 protein covalently. However, experimental methods are labour-intensive and cost-ineffective. In particular, in the post-genomic age, with the appearance of more and more genome data, these genome data are critical for both basic research and applications. Machine learning-based approaches are generally robust and effective in dealing with biological problems and may play a complementary role to wet experimental techniques. Therefore, it is very important to apply machine learning algorithm to recognize recombination hotspots.
During the past few years, some methods have been developed to identify recombination hotspots. Zhou et al. [12] firstly established a support vector machine (SVM)-based model to discriminate recombination hotspots from coldspots in S. cerevisiae by using codon composition. Subsequently, Jiang et al. [13] developed a new model based on gapped dinucleotide composition and random forest (RF) to predict meiotic recombination hotspots and coldspots in S. cerevisiae. Later on, Liu et al. [14] proposed an increment of diversity combined with quadratic discriminant (IDQD) model to predict recombination hotspots. In 2013, Chen et al. [15] designed a new DNA sample descriptor called pseudo dinucleotide composition (PseDNC) to improve the prediction accuracy [16]. Inspired by the concept of PseDNC, Li et al. [17] and Qiu et al. [18] also developed similar models to predict recombination hotspots, respectively. In 2014, Zhang and Liu [19] presented a model for predicting recombination hotspots based on the sequence and nucleosome occupancy in yeast. Subsequently, Liu et al. [20] incorporated the weight of features into the prediction model to identify recombination hotspots. To improve the performance, Dong et al. [21] combined the Z curve with PseDNC to distinguish recombination hotspots and coldspots. By fusing different modes of pseudo K-tuple nucleotide composition (PseKNC) and modes of dinucleotide-based autocross covariance into an ensemble classifier of clustering approach, another tool called iRSpot-EL was established [22]. In 2016, Kabir and Hayat [23] proposed a genetic algorithm-based ensemble model named iRSpot-GAEnsC for the discrimination between recombination hotspots and coldspots. In 2018, Zhang et al. constructed two models, namely iRSpot-ADPM [24] and iRSpot-PDI [25], to recognize recombination hotspots. Maruf and Shatabda [26] built a tool named iRSpot-SF for identifying recombination hotspots. More recently, Yang et al. [27] designed a powerful predictor called iRSpot-Pse6NC to identify recombination hotspots by incorporating the key hexamer compositions.
Although the models mentioned above reported good performances on recombination hotspots identification, they were trained and tested on different benchmark data sets, which prevent users from selecting the optimal model to perform predictions. Moreover, with the accumulation of more and more sequenced data, is there available objective and strict data that can be used to train a more powerful model? Thus, in this review, we provide a comprehensive survey of the most up-to-date progress of large-scale computational studies on recombination hotspots prediction. In total, we summarized the 15 methods listed in Table 1 in the following aspects, namely benchmark data set construction, feature extraction, prediction algorithm, performance evaluation strategies and software usability.
A comprehensive list of methods for prediction recombination hotspots of S. cerevisiae
| Methods | Web server | Data set | Feature extraction | Feature selection | Algorithm | Evaluation strategy |
|---|---|---|---|---|---|---|
| SVM-FCU | No | S1 | FCU | None | SVM-RBF | 10-fold CV |
| RF-DYMHC | Yes | S1, S2 | Gapped dinucleotide composition features | None | RF | 5-fold CV and Jackknife validation |
| IDQD-4-mer | No | S1, S2, S3 | 4-mer | None | IDQD | 5-fold CV |
| IRSpot-PseDNC | Yes | S1, S2, S3 | PseDNC | None | SVM-RBF | 5-fold CV and Jackknife validation |
| SVM-NACPseDNC | No | S1 | Nucleic acid composition (NAC), n-tier NAC and PseDNC | None | SVM-linear kernel | Jackknife validation |
| IRSpot-TNCPseAAC | Yes | S1, S2, S3 | TNC and PseAAC | None | SVM-RBF | Jackknife validation |
| Weighted features | No | S1 | k-mer, physical properties | None | Weighted features | 5-fold CV |
| IRSpot-EL | Yes | S2 | PseKNC, DACC | F-score | SVM-RBF | 5-fold CV |
| IDQD-Nu-occ | No | S1 | 4-mer, Nu-occ | None | IDQD | 5-fold CV |
| IRSpot-GAEnsC | No | S1 | TNC, DNC | None | ANN + SVM + RF | Jackknife validation |
| HcsPredictor | Yes | S1 | PseDNC+Z | None | LDM | 5-fold CV and Jackknife validation |
| IRSpot-ADPM | No | S1, S2 | ADPM | Wrapper SVM | SVM-RBF | 5-fold CV and Jackknife validation |
| IRSpot-PDI | No | S1, S2 | PDI | Wrapper SVM | SVM-RBF | Jackknife validation |
| IRSpot-SF | Yes | S2 | Gapped dinucleotide, TF-IDF, K-mer, Reverse K-mer | SVM-RFE | SVM-RBF | 5-fold CV and Jackknife validation |
| IRSpot-Pse6NC | Yes | S3 | Pse6NC | Binomial distribution | SVM-RBF | 5-fold CV |
| Methods | Web server | Data set | Feature extraction | Feature selection | Algorithm | Evaluation strategy |
|---|---|---|---|---|---|---|
| SVM-FCU | No | S1 | FCU | None | SVM-RBF | 10-fold CV |
| RF-DYMHC | Yes | S1, S2 | Gapped dinucleotide composition features | None | RF | 5-fold CV and Jackknife validation |
| IDQD-4-mer | No | S1, S2, S3 | 4-mer | None | IDQD | 5-fold CV |
| IRSpot-PseDNC | Yes | S1, S2, S3 | PseDNC | None | SVM-RBF | 5-fold CV and Jackknife validation |
| SVM-NACPseDNC | No | S1 | Nucleic acid composition (NAC), n-tier NAC and PseDNC | None | SVM-linear kernel | Jackknife validation |
| IRSpot-TNCPseAAC | Yes | S1, S2, S3 | TNC and PseAAC | None | SVM-RBF | Jackknife validation |
| Weighted features | No | S1 | k-mer, physical properties | None | Weighted features | 5-fold CV |
| IRSpot-EL | Yes | S2 | PseKNC, DACC | F-score | SVM-RBF | 5-fold CV |
| IDQD-Nu-occ | No | S1 | 4-mer, Nu-occ | None | IDQD | 5-fold CV |
| IRSpot-GAEnsC | No | S1 | TNC, DNC | None | ANN + SVM + RF | Jackknife validation |
| HcsPredictor | Yes | S1 | PseDNC+Z | None | LDM | 5-fold CV and Jackknife validation |
| IRSpot-ADPM | No | S1, S2 | ADPM | Wrapper SVM | SVM-RBF | 5-fold CV and Jackknife validation |
| IRSpot-PDI | No | S1, S2 | PDI | Wrapper SVM | SVM-RBF | Jackknife validation |
| IRSpot-SF | Yes | S2 | Gapped dinucleotide, TF-IDF, K-mer, Reverse K-mer | SVM-RFE | SVM-RBF | 5-fold CV and Jackknife validation |
| IRSpot-Pse6NC | Yes | S3 | Pse6NC | Binomial distribution | SVM-RBF | 5-fold CV |
A comprehensive list of methods for prediction recombination hotspots of S. cerevisiae
| Methods | Web server | Data set | Feature extraction | Feature selection | Algorithm | Evaluation strategy |
|---|---|---|---|---|---|---|
| SVM-FCU | No | S1 | FCU | None | SVM-RBF | 10-fold CV |
| RF-DYMHC | Yes | S1, S2 | Gapped dinucleotide composition features | None | RF | 5-fold CV and Jackknife validation |
| IDQD-4-mer | No | S1, S2, S3 | 4-mer | None | IDQD | 5-fold CV |
| IRSpot-PseDNC | Yes | S1, S2, S3 | PseDNC | None | SVM-RBF | 5-fold CV and Jackknife validation |
| SVM-NACPseDNC | No | S1 | Nucleic acid composition (NAC), n-tier NAC and PseDNC | None | SVM-linear kernel | Jackknife validation |
| IRSpot-TNCPseAAC | Yes | S1, S2, S3 | TNC and PseAAC | None | SVM-RBF | Jackknife validation |
| Weighted features | No | S1 | k-mer, physical properties | None | Weighted features | 5-fold CV |
| IRSpot-EL | Yes | S2 | PseKNC, DACC | F-score | SVM-RBF | 5-fold CV |
| IDQD-Nu-occ | No | S1 | 4-mer, Nu-occ | None | IDQD | 5-fold CV |
| IRSpot-GAEnsC | No | S1 | TNC, DNC | None | ANN + SVM + RF | Jackknife validation |
| HcsPredictor | Yes | S1 | PseDNC+Z | None | LDM | 5-fold CV and Jackknife validation |
| IRSpot-ADPM | No | S1, S2 | ADPM | Wrapper SVM | SVM-RBF | 5-fold CV and Jackknife validation |
| IRSpot-PDI | No | S1, S2 | PDI | Wrapper SVM | SVM-RBF | Jackknife validation |
| IRSpot-SF | Yes | S2 | Gapped dinucleotide, TF-IDF, K-mer, Reverse K-mer | SVM-RFE | SVM-RBF | 5-fold CV and Jackknife validation |
| IRSpot-Pse6NC | Yes | S3 | Pse6NC | Binomial distribution | SVM-RBF | 5-fold CV |
| Methods | Web server | Data set | Feature extraction | Feature selection | Algorithm | Evaluation strategy |
|---|---|---|---|---|---|---|
| SVM-FCU | No | S1 | FCU | None | SVM-RBF | 10-fold CV |
| RF-DYMHC | Yes | S1, S2 | Gapped dinucleotide composition features | None | RF | 5-fold CV and Jackknife validation |
| IDQD-4-mer | No | S1, S2, S3 | 4-mer | None | IDQD | 5-fold CV |
| IRSpot-PseDNC | Yes | S1, S2, S3 | PseDNC | None | SVM-RBF | 5-fold CV and Jackknife validation |
| SVM-NACPseDNC | No | S1 | Nucleic acid composition (NAC), n-tier NAC and PseDNC | None | SVM-linear kernel | Jackknife validation |
| IRSpot-TNCPseAAC | Yes | S1, S2, S3 | TNC and PseAAC | None | SVM-RBF | Jackknife validation |
| Weighted features | No | S1 | k-mer, physical properties | None | Weighted features | 5-fold CV |
| IRSpot-EL | Yes | S2 | PseKNC, DACC | F-score | SVM-RBF | 5-fold CV |
| IDQD-Nu-occ | No | S1 | 4-mer, Nu-occ | None | IDQD | 5-fold CV |
| IRSpot-GAEnsC | No | S1 | TNC, DNC | None | ANN + SVM + RF | Jackknife validation |
| HcsPredictor | Yes | S1 | PseDNC+Z | None | LDM | 5-fold CV and Jackknife validation |
| IRSpot-ADPM | No | S1, S2 | ADPM | Wrapper SVM | SVM-RBF | 5-fold CV and Jackknife validation |
| IRSpot-PDI | No | S1, S2 | PDI | Wrapper SVM | SVM-RBF | Jackknife validation |
| IRSpot-SF | Yes | S2 | Gapped dinucleotide, TF-IDF, K-mer, Reverse K-mer | SVM-RFE | SVM-RBF | 5-fold CV and Jackknife validation |
| IRSpot-Pse6NC | Yes | S3 | Pse6NC | Binomial distribution | SVM-RBF | 5-fold CV |
The recombination hotspots data sets of S. cerevisiae
The recombination hotspots data sets of S. cerevisiae
Despite the great progress made in the above research, there are still some problems. With the development of experimental technology, the resolution and sensitivity of genome-wide DSBs atlas have been greatly improved. However, the data set used in the previous models have not been improved. In previous data sets, due to the limitations of experimental methods, only DSBs within ORF can be found. However, the recombination hotspots are not limited to ORF, thus these models trained on such data set lack extensive application [11]. Furthermore, most of models did not pay attention to the problem of the sliding window size. They can only predict whether an input sequence contains recombination hotspots or not, but could not predict where the recombinant hotspots are located [27]. When a genome is scanned using a model, the question of how to set a window becomes more important.
To solve these problems, a new data set with high resolution and sensitivity was built to train a new predictor, in which the recombination hotspots are no longer limited to ORF [11]. The new model was examined on the chromosome XVI of S. cerevisiae. We believe that our work represents an important advancement in accelerating the discovery of recombination hotspots. We anticipate that our review will be helpful for future development of computational methods to efficiently and accurately identify recombination hotspots.
Methods and materials
The benchmark data sets
In 2000, Gerton et al. proposed the DNA microarray method to estimate the DSBs formation adjacent to each ORF in the S. cerevisiae at single-gene resolution. To estimate the DSBs formation adjacent to each ORF, they measured the hybridization ratio of a DSBs-enriched probe (P2) to a total genomic probe (P1) [1]. The relative strength of the recombination rate was estimated by the P2/P1 hybridization ratio. If the P2/P1 hybridization ratio ranked in the top 12.5% in more than five of seven experiments, an ORF was classified as ‘hot’, and if it ranked in the bottom 12.5%, it was classified as ‘cold’. Accordingly, the authors found 177 hotspots and 40 coldspots. In another study [13], based on Gerton et al.’s study, Jiang et al. reconstructed the data set and defined the relative hybridization ratio ≥1.5 as hotspots, while relative hybridization ratio <0.82 was defined as coldspots. Thus, Jiang et al. [13] obtained 490 hotspots and 591 coldspots, which form the training data set. The data set is called S1 and is available in Supplementary Material S1.
In Liu et al.’s study [22], to avoid redundancy and reduce homology deviations, sequences with similarity >75% were removed by using CD-HIT [28]. Then they obtained 478 hotspots and 572 coldspots, which formed data set S2 and is available in Supplementary Material S2.
It was found that the sequences in Supplementary Materials S1 and S2 are with different lengths, which prevented them from establishing a widely useful model because users do not know how long the length should be used for a query DNA sequence. Thus, an equal length data set was rebuilt [27]. The new data set, called S3, contains 490 recombination hotspots and 591 recombination coldspots with the length of 131 bp and is available in Supplementary Material S3.
The independent test data set contains the remaining 268 true recombination hotspots sequences and 280 non-recombination hotspots sequences in XVI chromosomes of S. cerevisiae, which are available in Supplementary Material S5.
Feature extraction
How to formulate a biological sequence with a vector is one of the most challenging problems in computational biology [30]. This is because nearly all the existing machine learning algorithms were developed to handle vectors rather than sequence samples [31]. To capture the key information of recombination hotspots, there are two types of feature encoding algorithms that have been proposed, namely ORF-based methods and non-ORF-based methods.
ORF-based approaches
Codon use frequency
Phase-specific PseDNC
Amino acid composition
where |${N}_{\mathrm{r}}$| is the number of the amino acid type |$r$| and |$N$| is the length of the sequence.
Trinucleotide composition and pseudo amino acid composition (TNCPseAAC)
where |${H}_n({A}_j)\ (n=1,2,\dots, 6)\ \mathrm{is}$| the standardized values of six physicochemical properties of amino acids. They are hydrophobicity, hydrophilicity, side-chain mass, pK1 (α-COOH), pK2 (NH3) and PI and can be obtained from Ref [18].
Non-ORF-based methods
Gapped dinucleotide composition features
K-mer composition
Reverse complement composition of k-mer
The reverse complement composition of k-mer is the normalized count of k-mer and its reverse complements present in the DNA sequence [26, 37]. The difference of this feature with k-mer composition is that the reverse complement composition of k-mer counts all the occurrences of the k-mer and its reverse complement.
Term frequency–inverse document frequency of k-mer
Pseudo K-tuple nucleotide composition
Weighted features
To quantify the importance of different features in prediction and incorporate different kinds of features in a proper way, Liu et al. [20] weighted the features using a variance-based feature selection method. First, samples were represented by possibly important features, such as k-mer distribution in sequences and sequence-order-related information. Second, weights of different features were calculated according to their ability to distinguish positive samples from negative samples. Third, values of the features were normalized via a standard conversion. Finally, samples were represented by weighted features (WF) by multiplying the normalized features with their corresponding weights.
Physical properties of DNA sequences
Associated dinucleotide product model (ADPM)
ADPM is an algorithm that uses an associated dinucleotide product model to define Chou’s pseudo components [24].
|${\varTheta}_{i,g,k,t}$|reflects the associated influence of dinucleotide pairs at different positions with genomic evolutionary. |${P}_{i,k}$| denotes the k-th property value of the i-th dinucleotide pair, which is composed of two adjacent nucleotides in the sequence. The given sequence can be converted into a feature vector with a uniform size of 15 × 15 ×|$g$|.
Property diversity information (PDI)
Dinucleotide-based auto-cross covariance
Dinucleotide-based auto-cross covariance (DACC) approach is a very special PseKNC mode, which is a combination of dinucleotide-based auto covariance and dinucleotide-based cross covariance [20].
Feature selection methods
As the dimension of the feature vector rises rapidly, we consider three issues: one is the large feature set that contains some redundant or irrelevant information; another is over-fitting, which results in low generalization ability of prediction model; the other is causing the curse of dimensionality and dyscalculia. It is necessary to employ feature selection process to improve these problems [38–40]. Three main directions have been developed for feature selection—filter, wrapper and embedded. Filters rely on the general characteristics of training data and carry out the feature selection process as a pre-processing step with independence of the induction algorithm. Wrappers involve optimizing a predictor as a part of the selection process. Embedded methods perform feature selection in the process of training and are usually specific to given learning machines. In our study, some works also use these three feature selection methods.
Binomial distribution
We should judge whether the occurrence of a particular feature is a stochastic event or not. According to the statistical theory, the binomial distribution was used to measure the occurrence probabilities of all features in positive samples and negative samples [27]. When the P value of a feature is small, the occurrence of the feature may not be a random event [41]. In this study, according to the statistical theory, the binomial distribution was used to measure the confidence of features. Then we use the incremental feature selection (IFS) to find out the best feature subset that could produce the maximum accuracy.
F-score
F-score is based on the idea that the differences within classes should be less than the differences between classes [42]. The larger the F-score value, the greater the difference between this feature and other features. It also indicates that the feature has stronger discrimination and greater effect on the prediction result. Specific implementation methods can be seen in a recent study [20].
Wrapper SVM
In this study, a wrapper feature selection process with SVM classifier is performed. Specifically, sequential forward selection strategy is adopted, in which features are sequentially added one by one to an empty candidate set until the addition of further features does not increase the criterion [25]. The criterion is defined by the overall accuracy obtained from SVM with n-fold cross-validation (CV). Finally, an optimal feature subset was constructed.
Recursive feature elimination for support vector machines
Recursive Feature Elimination for Support Vector Machines (SVM-RFE) was applied to select a group of important features [43]. This embedded method performs feature selection by iteratively training an SVM classifier with the current set of features and removing the least important feature indicated by the SVM [17, 26].
Model training
Support vector machine
Random forest
The RF algorithm is also a popular machine learning algorithm and has been successfully employed in dealing with various biological prediction problems [53–55]. RF is a classifier that contains multiple decision trees; it is adopted bagging and random feature selection strategy. In bagging, each tree is trained on a bootstrap sample of the training data, and predictions are made by the majority votes of trees. RF is a further development of bagging. Instead of using all features, RF randomly selects a subset of features to split at each node when growing a tree. To assess the prediction performance of the algorithm, RF performs a type of cross-validation in parallel with the training step by using the so-called out-of-bag samples [56]. The RF algorithm was implemented by the Random Forest R package [13].
Recently, Ke et al. [57] proposed a novel RF algorithm named LightGBM, which uses a novel gradient boosting decision tree algorithm, including gradient-based one-side sampling to extract relatively small number of samples according to gradient values and exclusive feature bundling to reduce the number of features.
Increment of diversity
Quadratic discriminant
In Liu’s study [60], a quadratic discriminant (QD) function based on Mahalanobis distance was used for prediction of recombination hotspots [60]. Mahalanobis distance is based on correlations between variables by which different patterns can be identified and analysed. It is a useful measure of difference between an unknown sample set and a known one. The combination of ID and QD algorithms led to a powerful classifier called IDQD.
Large margin distribution machine
The margin distribution has a crucial influence on the performance of classifiers. Large margin distribution machine (LDM), which is inspired by the generalization performance can be improved by optimizing the margin distribution through maximizing the margin mean and minimizing the margin variance simultaneously [21, 61]. Using LDM to implement classification generally requires two steps: first, mapping the feature vector to the high-dimensional space; second, using the hyperplane to maximize the margin mean while minimizing the margin variance, thereby conveniently separating the samples. There are two solvers in the LDM: the dual coordinate descent method and the average stochastic gradient descent method. In our study, we use the dual coordinate descent method.
Artificial neural network
Artificial neural network (ANN) abstracts the human brain neuron network from the perspective of information processing, establishes a simple model and forms different networks according to different connection methods. In the past decade or so, the research work of ANNs has been deepened and great progress has been made. It has been successfully applied in the field of bioinformatics [62, 63]. In this research [23], a variety of ANN methods have also been applied, such as probabilistic neural network, generalize regression neural network, feed forward neural network, fitting network and pattern recognition network.
K-nearest neighbour (KNN)
KNN is a widely used algorithm in the field of pattern recognition [23]. The flexibility of KNN is of great advantage for gene function classification problems, wherein the class boundaries are inherently vague, and many classes cannot be categorized by a simple model. The main idea of the classical KNN is the following: design a set of numerical features to describe each data point and select a metric, e.g. Euclidean distance, to measure the similarity of data points based on all features. Then for a target point, find its K closest points in the training samples based on the similarity metric and assign it to a class by the majority votes of its neighbours.
Comparison with state-of-the-art predictors on data sets S1
| Methods | 5-fold cross-validation | Jackknife validation | ||||||
|---|---|---|---|---|---|---|---|---|
| Sn(%) | Sp(%) | Acc(%) | MCC | Sn(%) | Sp(%) | Acc(%) | MCC | |
| RF-DYMHC | 80.59 | 84.26 | 82.05 | 0.638 | – | – | – | – |
| IDQD-4-mer | 79.40 | 81.00 | 80.30 | 0.603 | – | – | – | – |
| IRSpot-PseDNC | 81.63 | 88.14 | 85.19 | 0.692 | 73.06 | 89.49 | 82.04 | 0.638 |
| SVM-NACPseDNC | – | – | – | – | 76.12 | 90.69 | 84.09 | 0.680 |
| IRSpot-TNCPseAAC | – | – | – | – | 84.17 | 79.59 | 83.72 | 0.671 |
| Weighted-Features | 86.10 | 94.30 | 90.70 | 0.812 | – | – | – | – |
| IDQD-Nu-occ | 80.00 | 82.90 | 81.60 | 0.629 | – | – | – | – |
| IRSpot-GAEnsC | – | – | – | – | 80.08 | 88.07 | 84.46 | 0.690 |
| HcsPredictor | 77.82 | 89.78 | 84.36 | 0.685 | 78.78 | 90.69 | 85.29 | 0.704 |
| ANN-AAC | – | – | – | – | – | – | – | – |
| IRSpot-ADPM | – | – | – | – | 75.51 | 90.52 | 83.72 | 0.673 |
| IRSpot-PDI | – | – | – | – | 71.63 | 93.91 | 83.81 | 0.681 |
| Methods | 5-fold cross-validation | Jackknife validation | ||||||
|---|---|---|---|---|---|---|---|---|
| Sn(%) | Sp(%) | Acc(%) | MCC | Sn(%) | Sp(%) | Acc(%) | MCC | |
| RF-DYMHC | 80.59 | 84.26 | 82.05 | 0.638 | – | – | – | – |
| IDQD-4-mer | 79.40 | 81.00 | 80.30 | 0.603 | – | – | – | – |
| IRSpot-PseDNC | 81.63 | 88.14 | 85.19 | 0.692 | 73.06 | 89.49 | 82.04 | 0.638 |
| SVM-NACPseDNC | – | – | – | – | 76.12 | 90.69 | 84.09 | 0.680 |
| IRSpot-TNCPseAAC | – | – | – | – | 84.17 | 79.59 | 83.72 | 0.671 |
| Weighted-Features | 86.10 | 94.30 | 90.70 | 0.812 | – | – | – | – |
| IDQD-Nu-occ | 80.00 | 82.90 | 81.60 | 0.629 | – | – | – | – |
| IRSpot-GAEnsC | – | – | – | – | 80.08 | 88.07 | 84.46 | 0.690 |
| HcsPredictor | 77.82 | 89.78 | 84.36 | 0.685 | 78.78 | 90.69 | 85.29 | 0.704 |
| ANN-AAC | – | – | – | – | – | – | – | – |
| IRSpot-ADPM | – | – | – | – | 75.51 | 90.52 | 83.72 | 0.673 |
| IRSpot-PDI | – | – | – | – | 71.63 | 93.91 | 83.81 | 0.681 |
Comparison with state-of-the-art predictors on data sets S1
| Methods | 5-fold cross-validation | Jackknife validation | ||||||
|---|---|---|---|---|---|---|---|---|
| Sn(%) | Sp(%) | Acc(%) | MCC | Sn(%) | Sp(%) | Acc(%) | MCC | |
| RF-DYMHC | 80.59 | 84.26 | 82.05 | 0.638 | – | – | – | – |
| IDQD-4-mer | 79.40 | 81.00 | 80.30 | 0.603 | – | – | – | – |
| IRSpot-PseDNC | 81.63 | 88.14 | 85.19 | 0.692 | 73.06 | 89.49 | 82.04 | 0.638 |
| SVM-NACPseDNC | – | – | – | – | 76.12 | 90.69 | 84.09 | 0.680 |
| IRSpot-TNCPseAAC | – | – | – | – | 84.17 | 79.59 | 83.72 | 0.671 |
| Weighted-Features | 86.10 | 94.30 | 90.70 | 0.812 | – | – | – | – |
| IDQD-Nu-occ | 80.00 | 82.90 | 81.60 | 0.629 | – | – | – | – |
| IRSpot-GAEnsC | – | – | – | – | 80.08 | 88.07 | 84.46 | 0.690 |
| HcsPredictor | 77.82 | 89.78 | 84.36 | 0.685 | 78.78 | 90.69 | 85.29 | 0.704 |
| ANN-AAC | – | – | – | – | – | – | – | – |
| IRSpot-ADPM | – | – | – | – | 75.51 | 90.52 | 83.72 | 0.673 |
| IRSpot-PDI | – | – | – | – | 71.63 | 93.91 | 83.81 | 0.681 |
| Methods | 5-fold cross-validation | Jackknife validation | ||||||
|---|---|---|---|---|---|---|---|---|
| Sn(%) | Sp(%) | Acc(%) | MCC | Sn(%) | Sp(%) | Acc(%) | MCC | |
| RF-DYMHC | 80.59 | 84.26 | 82.05 | 0.638 | – | – | – | – |
| IDQD-4-mer | 79.40 | 81.00 | 80.30 | 0.603 | – | – | – | – |
| IRSpot-PseDNC | 81.63 | 88.14 | 85.19 | 0.692 | 73.06 | 89.49 | 82.04 | 0.638 |
| SVM-NACPseDNC | – | – | – | – | 76.12 | 90.69 | 84.09 | 0.680 |
| IRSpot-TNCPseAAC | – | – | – | – | 84.17 | 79.59 | 83.72 | 0.671 |
| Weighted-Features | 86.10 | 94.30 | 90.70 | 0.812 | – | – | – | – |
| IDQD-Nu-occ | 80.00 | 82.90 | 81.60 | 0.629 | – | – | – | – |
| IRSpot-GAEnsC | – | – | – | – | 80.08 | 88.07 | 84.46 | 0.690 |
| HcsPredictor | 77.82 | 89.78 | 84.36 | 0.685 | 78.78 | 90.69 | 85.29 | 0.704 |
| ANN-AAC | – | – | – | – | – | – | – | – |
| IRSpot-ADPM | – | – | – | – | 75.51 | 90.52 | 83.72 | 0.673 |
| IRSpot-PDI | – | – | – | – | 71.63 | 93.91 | 83.81 | 0.681 |
Performance evaluation
In order to measure the prediction quality, the following set of four metrics was used: sensitivity (Sn), specificity (Sp), overall accuracy (Acc) and Matthews correlation coefficient (MCC) [64–69].
Receiver operating characteristic curve
The receiver operating characteristic (ROC) curve [70–74] was also used to measure the prediction power of the current method, where its vertical coordinate is for the true positive rate (|$\mathrm{Sensitivity}$|) and the horizontal coordinate for the false positive rate (|$1-\mathrm{Specificity}$|). The area under the curve of ROC (AUC) of 0.5 is equivalent to random prediction, while a value of 1 represents a perfect one.
Comparison with state-of-the-art predictors on data sets S2
| Methods | 5-fold cross-validation | Jackknife cross-validation | ||||||
|---|---|---|---|---|---|---|---|---|
| Sn(%) | Sp(%) | Acc(%) | MCC | Sn(%) | Sp(%) | Acc(%) | MCC | |
| iRSpot-EL | 75.29 | 88.81 | 82.65 | 0.651 | – | – | – | – |
| RF-DYMHC | 73.01 | 86.56 | 80.40 | 0.605 | – | – | – | – |
| IDQD-4-mer | 79.52 | 81.82 | 80.77 | 0.616 | – | – | – | – |
| IRSpot-PseDNC | 71.75 | 85.84 | 79.33 | 0.583 | – | – | – | – |
| IRSpot-TNCPseAAC | 76.56 | 70.99 | 73.52 | 0.474 | – | – | – | – |
| IRSpot-ADPM | 77.19 | 90.73 | 84.57 | 0.690 | 76.36 | 90.56 | 84.10 | 0.681 |
| IRSpot-PDI | 71.84 | 92.95 | 83.16 | 0.666 | – | – | – | – |
| iRSpot-SF | 84.57 | 75.76 | 84.58 | 0.694 | 75.31 | 91.08 | 83.91 | 0.677 |
| Methods | 5-fold cross-validation | Jackknife cross-validation | ||||||
|---|---|---|---|---|---|---|---|---|
| Sn(%) | Sp(%) | Acc(%) | MCC | Sn(%) | Sp(%) | Acc(%) | MCC | |
| iRSpot-EL | 75.29 | 88.81 | 82.65 | 0.651 | – | – | – | – |
| RF-DYMHC | 73.01 | 86.56 | 80.40 | 0.605 | – | – | – | – |
| IDQD-4-mer | 79.52 | 81.82 | 80.77 | 0.616 | – | – | – | – |
| IRSpot-PseDNC | 71.75 | 85.84 | 79.33 | 0.583 | – | – | – | – |
| IRSpot-TNCPseAAC | 76.56 | 70.99 | 73.52 | 0.474 | – | – | – | – |
| IRSpot-ADPM | 77.19 | 90.73 | 84.57 | 0.690 | 76.36 | 90.56 | 84.10 | 0.681 |
| IRSpot-PDI | 71.84 | 92.95 | 83.16 | 0.666 | – | – | – | – |
| iRSpot-SF | 84.57 | 75.76 | 84.58 | 0.694 | 75.31 | 91.08 | 83.91 | 0.677 |
Comparison with state-of-the-art predictors on data sets S2
| Methods | 5-fold cross-validation | Jackknife cross-validation | ||||||
|---|---|---|---|---|---|---|---|---|
| Sn(%) | Sp(%) | Acc(%) | MCC | Sn(%) | Sp(%) | Acc(%) | MCC | |
| iRSpot-EL | 75.29 | 88.81 | 82.65 | 0.651 | – | – | – | – |
| RF-DYMHC | 73.01 | 86.56 | 80.40 | 0.605 | – | – | – | – |
| IDQD-4-mer | 79.52 | 81.82 | 80.77 | 0.616 | – | – | – | – |
| IRSpot-PseDNC | 71.75 | 85.84 | 79.33 | 0.583 | – | – | – | – |
| IRSpot-TNCPseAAC | 76.56 | 70.99 | 73.52 | 0.474 | – | – | – | – |
| IRSpot-ADPM | 77.19 | 90.73 | 84.57 | 0.690 | 76.36 | 90.56 | 84.10 | 0.681 |
| IRSpot-PDI | 71.84 | 92.95 | 83.16 | 0.666 | – | – | – | – |
| iRSpot-SF | 84.57 | 75.76 | 84.58 | 0.694 | 75.31 | 91.08 | 83.91 | 0.677 |
| Methods | 5-fold cross-validation | Jackknife cross-validation | ||||||
|---|---|---|---|---|---|---|---|---|
| Sn(%) | Sp(%) | Acc(%) | MCC | Sn(%) | Sp(%) | Acc(%) | MCC | |
| iRSpot-EL | 75.29 | 88.81 | 82.65 | 0.651 | – | – | – | – |
| RF-DYMHC | 73.01 | 86.56 | 80.40 | 0.605 | – | – | – | – |
| IDQD-4-mer | 79.52 | 81.82 | 80.77 | 0.616 | – | – | – | – |
| IRSpot-PseDNC | 71.75 | 85.84 | 79.33 | 0.583 | – | – | – | – |
| IRSpot-TNCPseAAC | 76.56 | 70.99 | 73.52 | 0.474 | – | – | – | – |
| IRSpot-ADPM | 77.19 | 90.73 | 84.57 | 0.690 | 76.36 | 90.56 | 84.10 | 0.681 |
| IRSpot-PDI | 71.84 | 92.95 | 83.16 | 0.666 | – | – | – | – |
| iRSpot-SF | 84.57 | 75.76 | 84.58 | 0.694 | 75.31 | 91.08 | 83.91 | 0.677 |
Model validation method
Cross-validation is a commonly used statistical analysis method that can be used to objectively evaluate the performance of a classification model [75, 76]. There are three commonly used cross-validation methods, namely independent data set test, n-fold cross-validation test and jackknife cross-validation test. To objectively evaluate the performance of different models, we tested them on the independent test data sets in this review.
Results and discussion
Comparison with state-of-the-art predictors on four data sets
Considering the fact that the existing methods were trained and tested based on different data, we evaluated them on data sets S1, S2, S3 and S4, respectively.
We firstly evaluated the 12 predictors that trained based on data set S1 and listed results in Table 3. Their results are based on the 5-fold cross-validation test and jackknife test. It can be seen that the model WF performs the best in 5-fold cross-validation test and got the best Sn/Sp/Acc/Mcc. WF is developed by using QD analysis based on WF. This result demonstrates that weighting the importance of the feature is effective for improving the accuracy of the model. In the jackknife test, the best result is obtained by the model of HcsPredictor. We also noticed that the model iRSpot-GAEnsC that integrates seven algorithms did not improve the prediction accuracy of the model. Therefore, we should cautiously choose the integration approach.
Eight predictors are based on the data set S2. As shown in Table 4, iRSpot-ADPM is the best predictor in 5-fold cross-validation. IRSpot-ADPM used the weights calculated by the SVM method to decrease features from 1575 to 85. After that, support vector machine-Gaussian radial basis function (SVM-RBF) is verified by 5-fold cross-validation and obtained an accuracy of 84.57%. Compared with the accuracy of 83.14% obtained by the iRSpot-ADPM1575, iRSpot-ADPM employed fewer features but obtained higher accuracy. So it can be understood that feature filtering is very helpful for building a model with a subset of non-redundant features.
Data set S3 is an isometric data set based on data set S1. Data set S3 can easily form a window to scan the entire sequence. This kind of predictor is more applicable, which is very important for discovering new recombination hotspots. As shown in Table 5, based on data set S3, the predictor iRSpot-Pse6NC achieved the best results.
Comparison with state-of-the-art predictors on data sets S3 in 5-fold cross-validation
| Methods | Sn(%) | Sp(%) | Acc(%) | MCC |
|---|---|---|---|---|
| IRSpot-Pse6NC | 75.71 | 91.03 | 84.08 | 0.6805 |
| IRSpot-PseDNC | 62.34 | 90.52 | 77.92 | 0.5585 |
| IRSpot-TNCPseAAC | 61.02 | 89.51 | 76.60 | 0.5334 |
| IDQD-4-mer | 69.59 | 75.09 | 72.59 | 0.4469 |
| Methods | Sn(%) | Sp(%) | Acc(%) | MCC |
|---|---|---|---|---|
| IRSpot-Pse6NC | 75.71 | 91.03 | 84.08 | 0.6805 |
| IRSpot-PseDNC | 62.34 | 90.52 | 77.92 | 0.5585 |
| IRSpot-TNCPseAAC | 61.02 | 89.51 | 76.60 | 0.5334 |
| IDQD-4-mer | 69.59 | 75.09 | 72.59 | 0.4469 |
Comparison with state-of-the-art predictors on data sets S3 in 5-fold cross-validation
| Methods | Sn(%) | Sp(%) | Acc(%) | MCC |
|---|---|---|---|---|
| IRSpot-Pse6NC | 75.71 | 91.03 | 84.08 | 0.6805 |
| IRSpot-PseDNC | 62.34 | 90.52 | 77.92 | 0.5585 |
| IRSpot-TNCPseAAC | 61.02 | 89.51 | 76.60 | 0.5334 |
| IDQD-4-mer | 69.59 | 75.09 | 72.59 | 0.4469 |
| Methods | Sn(%) | Sp(%) | Acc(%) | MCC |
|---|---|---|---|---|
| IRSpot-Pse6NC | 75.71 | 91.03 | 84.08 | 0.6805 |
| IRSpot-PseDNC | 62.34 | 90.52 | 77.92 | 0.5585 |
| IRSpot-TNCPseAAC | 61.02 | 89.51 | 76.60 | 0.5334 |
| IDQD-4-mer | 69.59 | 75.09 | 72.59 | 0.4469 |
Based on data set S4 containing both ORF and non-ORF recombination hotspots, we construct a predictor called iRSpot-Pse6NC2.0 by incorporating the key hexamer features into the general PseKNC via the binomial distribution feature selection approach. We sorted the features according to the scores obtained from binomial distribution. By using overall accuracy as vertical coordinates and feature numbers as horizontal coordinates, we plotted IFS curve as shown in Figure 1. It was noticed that the peak of the curve is 77.61%, which is located at horizontal coordinate of 1000. This result (77.61%) is higher than that (74.85%) by using all features. Accordingly, the 1000 hexamers were selected to form the optimal feature subset to train the prediction model.
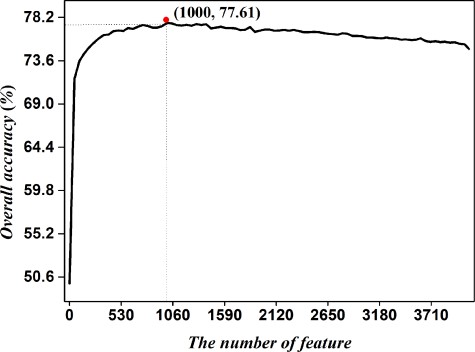
IFS curve for predicting recombination hotspots based on the 5-fold cross-validation. An IFS peak of 77.61% was observed when using the top 1000 hexamers to perform prediction
To further investigate the performance of the iRSpot-Pse6NC2.0, we drew the ROC curve in Figure 2. It shows that the AUC reaches a value of 0.833, indicating that the proposed method is quite promising and holds very high potential to become a useful high-throughput tool for predicting recombination hotspots.
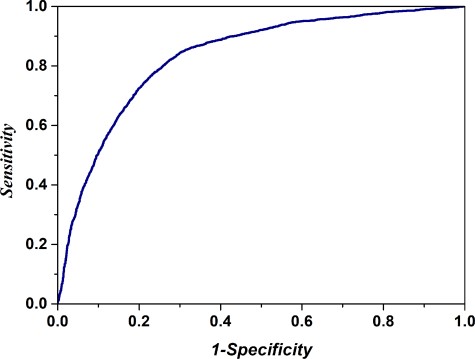
The ROC curve for identifying recombination hotspots by using 1000 optimal hexamers. The AUC of 0.833 was obtained in 5-fold cross-validation. The diagonal dot line denotes a random guess with the AUC of 0.5
Comparison with physical properties
In S. cerevisiae, both proteins Set1 and Spo11 affect the formation of DSBs through specific binding of DNA sequences [77, 78]. As a sequence feature, the hexamer can reflect the influence of these two proteins on the formation of DSBs. Due to nucleosome occupancy displaying an important role in the DSBs, we further investigated the influence of nucleosome occupancy on recombination spots prediction by adding feature of physical properties of nucleosome [20]. As it can be seen from Table 6, the nucleosome feature could produce the accuracy of 74.74%, demonstrating that the nucleosomes could influence the recombination. However, by combining the nucleosome feature with optimal hexamer composition, the accuracy reduced to 74.20%, suggesting that there is information redundancy. In fact, many studies have shown that hexamer composition is an important feature that could describe DNA motifs and could be utilized to identify a variety of DNA regulatory elements [77, 79]. Our results exactly reflected this property. The maximum accuracy of 77.61% was obtained by the optimal hexamers.
Comparison with physical properties on data set S4
| Feature | Algorithm | 5-fold cross-validation | ||
|---|---|---|---|---|
| Sn(%) | Sp(%) | Acc(%) | ||
| Optimal hexamers | SVM | 75.24 | 78.01 | 77.611 |
| Optimal hexamers | LightGBM | 77.24 | 75.80 | 76.531 |
| Physical properties | SVM | 72.91 | 74.89 | 74.742 |
| Optimal hexamers & Physical properties | SVM | 73.59 | 74.79 | 74.197 |
| Feature | Algorithm | 5-fold cross-validation | ||
|---|---|---|---|---|
| Sn(%) | Sp(%) | Acc(%) | ||
| Optimal hexamers | SVM | 75.24 | 78.01 | 77.611 |
| Optimal hexamers | LightGBM | 77.24 | 75.80 | 76.531 |
| Physical properties | SVM | 72.91 | 74.89 | 74.742 |
| Optimal hexamers & Physical properties | SVM | 73.59 | 74.79 | 74.197 |
Comparison with physical properties on data set S4
| Feature | Algorithm | 5-fold cross-validation | ||
|---|---|---|---|---|
| Sn(%) | Sp(%) | Acc(%) | ||
| Optimal hexamers | SVM | 75.24 | 78.01 | 77.611 |
| Optimal hexamers | LightGBM | 77.24 | 75.80 | 76.531 |
| Physical properties | SVM | 72.91 | 74.89 | 74.742 |
| Optimal hexamers & Physical properties | SVM | 73.59 | 74.79 | 74.197 |
| Feature | Algorithm | 5-fold cross-validation | ||
|---|---|---|---|---|
| Sn(%) | Sp(%) | Acc(%) | ||
| Optimal hexamers | SVM | 75.24 | 78.01 | 77.611 |
| Optimal hexamers | LightGBM | 77.24 | 75.80 | 76.531 |
| Physical properties | SVM | 72.91 | 74.89 | 74.742 |
| Optimal hexamers & Physical properties | SVM | 73.59 | 74.79 | 74.197 |
We also analysed the top 50 hexamer features that had the greatest impact on the results and found that the proportion of AT-content in these hexamers were relatively high. In S. cerevisiae, although recombination hotspots and GC-content at longer ranges were positively correlated, studies have found that many recombination spots are mostly in intergenic regions, which tend to be more at AT-rich than their surrounding regions [11].
Availability of online web server
For the convenience of the vast majority of experimental scientists, an online service is usually built. Users only need to submit the data in FASTA format to complete the prediction. Among the 15 existing predictors, only seven of them provide the online service. After checking them, as shown in Table 7, we found that only four web servers can be used at present.
The URL addresses for the listed tools
| Methods | Webservers | Available or not |
|---|---|---|
| iRSpotPseDNC | http://lin-group.cn/server/iRSpot-PseDNC | Yes |
| iRSpot-pse6NC | http://lin-group.cn/server/iRSpot-pse6NC/ | Yes |
| iRSpot-SF | http://irspot.pythonanywhere.com/server | No |
| RF-DYMHC | http://www.bioinf.seu.edu.cn/Recombination/ | No |
| HcsPredictor | http://cefg.cn/HcsPredictor | No |
| iRSpot-EL | http://bioinformatics.hitsz.edu.cn/iRSpot-EL/ | Yes |
| iRSpot-TNCPseAAC | http://www.jci-bioinfo.cn/iRSpot-TNCPseAAC | Yes |
| Methods | Webservers | Available or not |
|---|---|---|
| iRSpotPseDNC | http://lin-group.cn/server/iRSpot-PseDNC | Yes |
| iRSpot-pse6NC | http://lin-group.cn/server/iRSpot-pse6NC/ | Yes |
| iRSpot-SF | http://irspot.pythonanywhere.com/server | No |
| RF-DYMHC | http://www.bioinf.seu.edu.cn/Recombination/ | No |
| HcsPredictor | http://cefg.cn/HcsPredictor | No |
| iRSpot-EL | http://bioinformatics.hitsz.edu.cn/iRSpot-EL/ | Yes |
| iRSpot-TNCPseAAC | http://www.jci-bioinfo.cn/iRSpot-TNCPseAAC | Yes |
The URL addresses for the listed tools
| Methods | Webservers | Available or not |
|---|---|---|
| iRSpotPseDNC | http://lin-group.cn/server/iRSpot-PseDNC | Yes |
| iRSpot-pse6NC | http://lin-group.cn/server/iRSpot-pse6NC/ | Yes |
| iRSpot-SF | http://irspot.pythonanywhere.com/server | No |
| RF-DYMHC | http://www.bioinf.seu.edu.cn/Recombination/ | No |
| HcsPredictor | http://cefg.cn/HcsPredictor | No |
| iRSpot-EL | http://bioinformatics.hitsz.edu.cn/iRSpot-EL/ | Yes |
| iRSpot-TNCPseAAC | http://www.jci-bioinfo.cn/iRSpot-TNCPseAAC | Yes |
| Methods | Webservers | Available or not |
|---|---|---|
| iRSpotPseDNC | http://lin-group.cn/server/iRSpot-PseDNC | Yes |
| iRSpot-pse6NC | http://lin-group.cn/server/iRSpot-pse6NC/ | Yes |
| iRSpot-SF | http://irspot.pythonanywhere.com/server | No |
| RF-DYMHC | http://www.bioinf.seu.edu.cn/Recombination/ | No |
| HcsPredictor | http://cefg.cn/HcsPredictor | No |
| iRSpot-EL | http://bioinformatics.hitsz.edu.cn/iRSpot-EL/ | Yes |
| iRSpot-TNCPseAAC | http://www.jci-bioinfo.cn/iRSpot-TNCPseAAC | Yes |
Performance comparison on the yeast chromosome XVI
If the input sequence is too long, there will be two problems. One is that it cannot achieve a good prediction effect; the other is that it cannot predict whether the sequence contains multiple recombinant hotspots. In order to solve this problem, some predictors introduce sliding window method, which is to use sliding window scanning the input sequences and then predict the sub-sequence. There are two kinds of methods for the implementation of sliding window. One is iRSpot-Pse6NC, where in the model is built from the equal-length data set. When the prediction is made, the input sequence is divided into sub-sequences with the same length as that in the training data set. Another is iRSpot-EL. In the predictor, the input sequence is segmented according to the length of the window set by the user, and then the generated sub-sequence is predicted. Both methods have their own advantages. iRSpot-Pse6NC determines the most appropriate window length, while iRSpot-EL retains the complete experimental data as the training set. In newly constructed predictor iRSpot-Pse6NC2.0, we combined the above two advantages, retained complete experimental data to construct the training set and took the average length of sequence as the length of sliding window (189 bp).
Then, we used the yeast chromosome XVI to compare the performance of the predictors. We submitted the yeast chromosome XVI in FASTA format to the web servers listed in Table 7 and iRSpot-Pse6NC2.0 to obtain corresponding prediction results.
The predictor iRSpot-PseDNC and iRSpot-TNCPseAAC could not predict chromosome XVI directly, because the whole chromosome sequence is too long to be predicted. The predictor iRSpot-EL cannot be implemented according to the preset parameters. The predictor iRSpot-pse6NC segmented the chromosome according to the window length of 131 bp to generate sub-sequence and then obtained the results of sub-sequence. iRSpot-Pse6NC2.0 segmented the chromosome in a window length of 189 bp with the step of 189 bp. Then, we analysed the prediction results of iRSpot-Pse6NC and iRSpot-Pse6NC2.0. As shown in Figure 3, the average result of iRSpot-Pse6NC2.0 (Figure 3A) is higher than iRSpot-Pse6NC(Figure 3B), indicating that it is important to accurately predict the recombination hotspot region to retain the complete experimental data as the training set.

The prediction accuracy of (A) iRSpot-Pse6NC2.0 and (B) iRSpot-Pse6NC on the chromosome XVI.
In order to provide a detail analysis, we sorted the prediction probability of the 268 recombination hotspots that have been experimentally confirmed in a descending order. Then we used each prediction probability values as a threshold and calculated the number of sub-sequences with an accuracy greater than the threshold; the detailed results are shown in Figure 4. From Figures 3A and 4A, 220 experimentally verified recombination hotspots were correctly identified with an accuracy of 82.10% (220/268) and 1111 additional predictions were obtained by iRSpot-Pse6NC2.0. As indicated in Figures 3B and 4B, 109 experimentally verified recombination hotspots were correctly identified with an accuracy of 40.67% (109/268) and 1084 additional predictions were obtained by iRSpot-Pse6NC. These results demonstrate that the iRSpot-Pse6NC2.0 is more credible, reliable and robust for finding potential recombination hotspots.
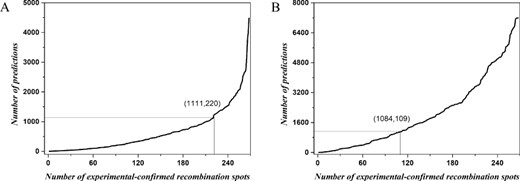
Prediction results of (A) iRSpot-Pse6NC2.0 and (B) iRSpot-Pse6NC on the chromosome XVI.
Performance analysis on the independent data set
We further compared the prediction performance of our proposed iRSpot-Pse6NC2.0 with that of iRSpot-PseDNC and iRSpot-TNCPseAAC based on the independent data set. The comparative results were listed in Table 8. The accuracy of iRSpot-PseDNC and iRSpot-TNCPseAAC was 56.16% and 54.58%,respectively, which is lower than 77.43% obtained by iRSpot-Pse6NC2.0, indicating that iRSpot-Pse6NC2.0 is better than iRSpot-TNCPseAAC and iRSpotPseDNC. This may be due to the fact that iRSpot-TNCPseAAC and iRSpotPseDNC were trained based on ORF data, while the data set of iRSpot-Pse6NC2.0 were constructed based on the data derived from the entire chromosome. The hexamer feature used in iRSpot-Pse6NC2.0 was obtained from DNA sequences directly. Thus, the model is more flexible.
Comparison with state-of-the-art predictors on independent data set S4
| Methods | Sn(%) | Sp(%) | Acc(%) |
|---|---|---|---|
| iRSpot-PseDNC | 76.86 | 35.44 | 56.16 |
| iRSpot-TNCPseAAC | 56.34 | 52.61 | 54.48 |
| iRSpot-Pse6NC2.0 | 77.23 | 77.61 | 77.43 |
| Methods | Sn(%) | Sp(%) | Acc(%) |
|---|---|---|---|
| iRSpot-PseDNC | 76.86 | 35.44 | 56.16 |
| iRSpot-TNCPseAAC | 56.34 | 52.61 | 54.48 |
| iRSpot-Pse6NC2.0 | 77.23 | 77.61 | 77.43 |
Comparison with state-of-the-art predictors on independent data set S4
| Methods | Sn(%) | Sp(%) | Acc(%) |
|---|---|---|---|
| iRSpot-PseDNC | 76.86 | 35.44 | 56.16 |
| iRSpot-TNCPseAAC | 56.34 | 52.61 | 54.48 |
| iRSpot-Pse6NC2.0 | 77.23 | 77.61 | 77.43 |
| Methods | Sn(%) | Sp(%) | Acc(%) |
|---|---|---|---|
| iRSpot-PseDNC | 76.86 | 35.44 | 56.16 |
| iRSpot-TNCPseAAC | 56.34 | 52.61 | 54.48 |
| iRSpot-Pse6NC2.0 | 77.23 | 77.61 | 77.43 |
Conclusion
Meiotic recombination has important roles in genome diversity and evolution, which is one of the most important driving forces of evolution. Investigations on recombination events and especially identification of recombination hotspots are significant for understanding the mechanism of recombination initiation. Since the drawbacks of experimental methods, computational methods will assist to identify recombination hotspots in a more efficient way. In this work, we have introduced and comprehensively evaluated the currently available tools for the prediction of recombination hotspots in S. cerevisiae. These computational methods were discussed and compared in terms of underlying algorithms, extracted features, predictive capability and practical utility. Subsequently, a new predictor called iRSpot-Pse6NC2.0 was constructed based on a more objective benchmark data set. To obtain a more objective performance evaluation, we constructed an independent test data set to benchmark all tools. The results demonstrated that the new predictor iRSpot-Pse6NC2.0 is superior to existing tools in the identification of recombination hotspots. We anticipate that iRSpot-Pse6NC2.0 available at http://lin-group.cn/server/iRSpot-Pse6NC2.0 will become a powerful tool for identifying recombination hotspots.
It should be noted that the proposed model iRSpot-Pse6NC2.0 was constructed based on a single-cell organism (S. cerevisiae) data. Due to species specificity, it may not be suitable for higher eukaryotes. However, the model can be used in other fungal genomes. In fact, species specificity is an important factor affecting the prediction performance of proposed models, so more and more bioinformatics models have been constructed for species specificity prediction [80, 81]. In the future, with the accumulation of higher eukaryotes data, we will update our model, which will be suitable for more species.
A total of 15 computational methods developed for identifying recombination hotspots were comprehensively summarized and discussed.
Several comparisons were performed to investigate the predictive capability of published methods.
A high-quality data set was built to train and test a new prediction model for identifying recombination hotspots.
A user-friendly web server was developed to recognize recombination hotspots.
Funding
This work was supported by the National Nature Scientific Foundation of China (61772119, 31771471, 61861036) and the Science Strength Promotion Programme of UESTC.
Hui Yang is a PhD candidate at the Center for Informational Biology, University of Electronic Science and Technology of China. Her research interests are bioinformatics, machine learning, RNA modification site and recombination spots.
Wuritu Yang is an associate professor at the State Key Laboratory of Reproductive Regulation and Breeding of Grassland Livestock, Inner Mongolia University. His research is in the areas of bioinformatics.
Fu-Ying Dao is a PhD candidate at the Center for Informational Biology, University of Electronic Science and Technology of China. Her research interests are bioinformatics, machine learning and DNA replication regulation.
Hao Lv is a PhD candidate at the Center for Informational Biology, University of Electronic Science and Technology of China. Her research interests are bioinformatics, machine learning and DNA and RNA modification site.
Hui Ding is an associate professor at the Center for Informational Biology, University of Electronic Science and Technology of China. Her research is in the areas of computational system biology.
Wei Chen is a professor at the Innovative Institute of Chinese Medicine and Pharmacy, Chengdu University of Traditional Chinese Medicine. His research is in the areas of bioinformatics, computational epigenetics and epitranscriptome.
Hao Lin is a professor at the Center for Informational Biology, University of Electronic Science and Technology of China. His research is in the areas of bioinformatics and system biology.

{kind=link}
{kind=link}
{kind=link}
{kind=link}