




Abstract
Computational detection methods have been widely used in studies on the biogenesis and the function of circular RNAs (circRNAs). However, all of the existing tools showed disadvantages on certain aspects of circRNA detection. Here, we propose an improved multithreading detection tool, CIRI2, which used an adapted maximum likelihood estimation based on multiple seed matching to identify back-spliced junction reads and to filter false positives derived from repetitive sequences and mapping errors. We established objective assessment criteria based on real data from RNase R-treated samples and systematically compared 10 circular detection tools, which demonstrated that CIRI2 outperformed its previous version CIRI and all other widely used tools, featured with remarkably balanced sensitivity, reliability, duration and RAM usage.
Introduction
As a new class of noncoding RNAs, more and more circular RNAs (circRNAs) have been found in diverse cell types, cellular components, developmental stages and eukaryotic organisms [1–6]. Recent studies have also pointed to distinct biogenesis mechanisms and functions of circRNAs. For example, in contrast with several known circular microRNA sponges discovered in cytoplasm [7, 8], nucleus-localized EIciRNAs were found to function as parental gene transcription activators [9]. In addition, the main circularization mechanism in yeast was reported to be exon-containing lariat mediation [10] instead of the prevalent complementary intron mediation in multicellular organisms [11–14]. A recent study revealed that more than one-third of abundant circRNAs during epithelial–mesenchymal transition were promoted by RNA-binding protein QKI [15] rather than the above two mechanisms.
Currently existing circRNA detection methods are all based on identification of back-spliced junction (BSJ) reads, and they can be further divided into annotation-dependent (such as MapSplice [16], CIRCexplorer [11] and KNIFE [17]) and de novo (such as find_circ [8], segemehl [18] and CIRI [19]) algorithms. These methods are not only used for a direct investigation of circRNA loci but also as basis of more complicated analyses. For example, our previous method CIRI facilitated the development of another algorithm on characterizing internal structure and alternative splicing within circRNAs by providing BSJs and corresponding reads [20]. Additionally, analysis of microRNA target, circRNA differential expression and parental gene enrichment all unavoidably call for more sensitive and reliable circRNA detection algorithm. However, current methods still have room for improvement. In a recent report, Hansen et al. [21] pointed out that prediction outputs of several detection algorithms were largely distinct with each other, and all showed disadvantages on specific aspects of performance. Such disadvantages were reflected in low sensitivity, low reliability, long duration, high RAM usage and/or complicated pipeline, which indeed resulted from the complexities of eukaryotic transcription and splicing as well as differential expression of circRNAs from various origins. For example, our previous method CIRI had the highest sensitivity of all methods, which is essential for unbiased and in-depth study of circRNA, but such comprehensive detection may lead to relatively high false discovery rate (FDR).
Here, we propose CIRI2, a program designed to differentiate BSJ reads from non-BSJ reads using efficient maximum likelihood estimation (MLE) based on multiple seed matching. Such MLE helps CIRI2 differentiate BSJ reads from non-BSJ reads with erroneous mapping to repetitive sequence, and thus achieve balanced performance with both low FDR and high sensitivity. This increased differentiation ability combined with multithreading allows CIRI2 to run faster and more efficiently use RAM. Through comprehensive evaluation, we demonstrated the advantages of CIRI2 compared with CIRI and all other eight algorithms.
Materials and methods
Overview of CIRI2
CIRI2 has the same input requirement as CIRI, including FASTA formatted reference sequences and the SAM alignment generated by BWA-MEM [22], for de novo detection of circRNAs based on BSJs. An optional GTF input can also be used by CIRI2 for an extra annotation-dependent detection according to known linear RNA exon boundaries, as well as for detailed annotation of all circRNA loci. CIRI2 is extensively optimized for the key steps in circRNA detection, including inferring original region for sequencing read segments based on seed matching, and distinguishing BSJ reads from forward-spliced junction (FSJ) reads based on an adapted MLE. CIRI2 is applicable to sequencing data with mixed read lengths, and can be run with multiple threads. CIRI2 is freely available together with full documentation at https://sourceforge.net/projects/ciri/.
Determination of the segment location by multiple seed matching
There are situations where an aligner cannot determine the mapping position of a segment in a sequencing read [23, 24], but it is often crucial for a circRNA detection tool to infer whether the segment is from a given genomic region. In CIRI, certain BSJ reads with a short segment flanking the BSJ that cannot be precisely aligned to the reference sequence by short-read mappers, named as ‘unbalanced junction reads’, are detected in the second round scanning. Such short segments are positioned by dynamic programming alignments with a group of segments from other reads with confident mapping positions (Supplementary Figure S1). These dynamic programming alignments are often computing-intensive. In contrast, CIRI2 can quickly achieve this key step by seed matching. If the segment is from a given genomic region, all of the seeds from the segment should be found in this genomic region, with the exception of sequencing error or interrupting intron. In detail, if only considering sequencing error rate i, for a seed with length of m, the probability that the seed is found in its genomic region can be simply estimated as (1-i)m, whereas the probability of a seed to be randomly found in any genomic region with length l can be estimated as (l-m + 1)/4m. Thus, a quick match of a seed in a given genomic region can be used to infer whether the original segment of the seed is from the genomic region when a proper m can be chosen to keep the sensitivity (1-i)m high and simultaneously to remain the FDR (l-m + 1)/4m low.
Considering that short seeds are more likely to cause spurious matches, the above inference depending on single seed matching often results in high occasionality. A much more robust solution is to apply multiple seed matching to determine the location of a segment, where a majority of seeds should perfectly match their original genomic location while a small fraction may fail to match because of sequencing errors or interrupting introns. In detail, CIRI2 first divides the segment into n seeds with length m and attempts to find the location of each seed in a given genomic region with the same length l. If only considering sequencing error rate i, the number of seeds (k) that are found in its genomic region follows a binomial distribution B(n, (1-i)m), whereas the number of seeds (k’) that are randomly found in any other genomic region follows a binomial distribution B(n, (l-m + 1)/4m). For example, for a 50 bp segment divided into five seeds with length of 10 bp, it will have 4.52±0.66 seeds perfectly matched to its genomic region with a sequencing error rate at 1%, and have 0.48 ± 0.66 seeds randomly matched to other regions within a length of 100 kb.
Detection of BSJ reads based on MLE
The dynamic programming alignment implemented in CIRI is dependent on the balanced BSJ reads directly indicated by BWA-MEM. Consequently, falsely reported balanced BSJ reads caused by spurious read mapping may lead to incorrect circRNA predictions. In CIRI2, however, the detection of unbalanced BSJ reads, as well as the balanced BSJ reads with low mapping quality, is more cautious. An adapted MLE is designed for such detection by choosing one of the two possible genomic regions of the key segment in these reads, according to the matched seed numbers in these two regions that follow binomial distributions with different probabilities.
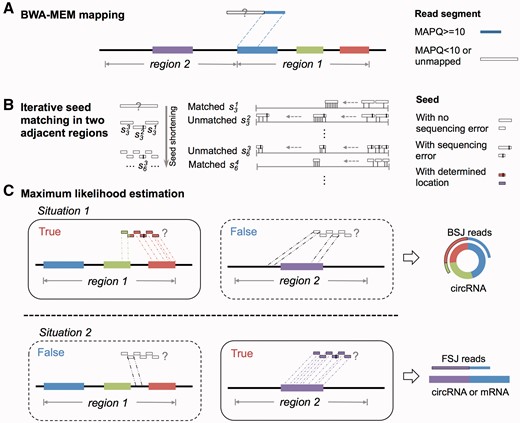
The MLE-based algorithm implemented in CIRI2. (A) Detection of back-spliced read can be simplified as making a decision on whether the specific segment is from genomic Region 1 (BSJ) or Region 2 (FSJ). (B) CIRI2 divides the segment into seeds, and attempts to perfectly match the seeds in Region 1 and Region 2. (C) A MLE is adopted to position the segment based on seed matching.
For example, for a 50 bp segment divided into five seeds with length of 10 bp, the overall FDR of the above MLE is about 0.01% in theory when the sequencing error rate is 1% and both lengths of the two regions are 100 kb. Although the above estimation of FDR is based on random sequence, the MLE model is also effective on avoiding false prediction in repetitive regions. As shown in Supplementary Figure S2, the model directly compares matched seeds within the two candidate regions of repetitive sequence, and, thus, subtle differences between the two regions can be easily detected.
The above MLE is also used in paired-end mapping filtration for all candidate BSJ reads. In the actual implementation of CIRI2, we made several adjustments to improve the efficiency of the above MLE: (1) set the minimum and maximum of l as 50 and 200 kb, respectively, so that the selected genomic regions can cover the vast majority of nearby BSJ and FSJ while simultaneously save running time; (2) use overlapped seeds instead of independent seeds to maximize the utility of segment sequence; (3) set additional matching thresholds and consider relative positions of matched seeds to further decrease FDR for BSJ read detection, especially for short seeds; and (4) adopt an iterative seed matching in a descending order of length m, in which shorter seeds with higher sensitivity are used in case that longer seeds fail to achieve the matching thresholds.
The implementation of multithreading in CIRI2
To facilitate large data set analysis, multithreading is implemented in CIRI2 using Perl module ‘threads’. When user designates more than one thread by parameter T, CIRI2 will first divide the SAM file into the corresponding number of equal parts. Because such division will cause separation of alignment records from the same reads at division points, CIRI2 records these reads and subsequently extracts their remaining alignments from the next part and add them back to make sure every divided part of SAM file has the complete alignment record for read pairs. During the double scanning of SAM records, CIRI2 allocates threads to each of the divided part, and then detects BSJ reads based on the MLE as described above. It should be noted that the ‘threads’ module will increase RAM usage for each allocated thread. To reduce RAM usage, CIRI2 stores intermediate results (i.e. candidate BSJs and the corresponding reads) into a temporary file when scanning the SAM alignment. After all threads get finished, these temporary files will be processed by one thread for final circRNA prediction.
Benchmarking circRNA detection using 10 algorithms
RNA-seq data sets generated in previous studies were used for circRNA detection by 10 popular tools, including CIRI [19], CIRI2, CIRCexplorer [11], circRNA_finder [1], DCC [25], find_circ [8], KNIFE [17], MapSplice [16], segemehl [18] and UROBORUS [26], with the parameters shown in Supplementary File 1, which were suggested by algorithm developers or previous studies. Four data sets of Hs68 cell lines [27], including two with RNase R treatment (accession numbers SRR444974 and SRR445016) and two without RNase R treatment (accession numbers SRR444655 and SRR444975), were downloaded from the SRA database. Two data sets of HEK293 cell lines [20], including one with RNase R treatment (accession number SRR3479244) and one without RNase R treatment (accession number SRR3479243), were also downloaded from the SRA database. All algorithms were run for each individual data set, and BSJ read counts for each circRNA in the two replicates with the same treatment of Hs68 cell line were summed. Human hg19 genome sequences were downloaded from the UCSC Web site and used as references. Duration and RAM usage were monitored by running command ‘qstat –f Job_ID’ on our Portable Batch System every 10 s for each detection.
Calculation of sensitivity, FDR and F1 score
We used a similar criteria with a previous study [21] to evaluate candidate circRNAs detected with BSJ reads count ≥3 in the data sets without RNase R treatment. In detail, if candidate circRNAs detected by each tool were obviously enriched after RNase R treatment (at least 3-fold increase of BSJ reads count), they were labeled as true positives. In contrast, candidate circRNAs not detected or largely depleted (with fewer BSJ reads count) after RNase R treatment were labeled as false positives. The remaining candidate circRNAs detected after RNase R treatment but without obvious enrichment were labeled as undetermined. The ratio of false positives in all of the predictions by each tool was calculated as FDR of the algorithm. We next summed true positives predicted by all tools as the estimated total circRNAs in the data sets. The ratio of true positives detected by each tool in the above total circRNAs was used to evaluate the sensitivity of the tool. To compare performances of all tools, we applied a single metric F1 score that simultaneously considers sensitivity and FDR of the tool. The F1 score was calculated according to the formula F1 = 2*sensitivity*(1-FDR)/(sensitivity + 1-FDR).
Results
Improved circular identification for low FDR and low time complexity
To quantify the performance improvement of CIRI2 compared with CIRI, we applied both tools to previously generated RNA-seq data sets of Hs68 [27] without RNase R treatment. As shown in Figure 2A, predictions of CIRI and CIRI2 in the data sets showed a significant overlap—3041 of 4037 candidate circRNAs in CIRI were also detected by CIRI2. The remaining 996 candidates were filtered out by CIRI2, and there were also 148 candidates predicted by CIRI2 but not by CIRI. We next determined the number of true-positive and false-positive circRNAs based on BSJ read enrichment by incorporating the data sets treated with RNase R. As shown in Figure 2B, CIRI2 could effectively remove false positives in CIRI (435 versus 1062) but kept true positives largely unchanged (2440 versus 2594), which indicated the balanced performance of CIRI2 on both FDR and sensitivity. Moreover, the vast majority of lost true positives had lower expression levels, but the removed false positives were mainly composed of abundant predictions. When focusing on the 996 and 148 predictions exclusively detected by the two algorithms, we found that CIRI2 only missed 33 true positives with more than five BSJ reads, but it successfully filtered 325 false positives more than CIRI at the same expression level (Figure 2C). When focusing on the top 1000 most abundant predictions, we observed a more stable reliability of CIRI2 compared with CIRI (Figure 2D). We also tested them on another data set of HEK293 with longer sequencing reads, and found a similar trend (Supplementary Figure S3).
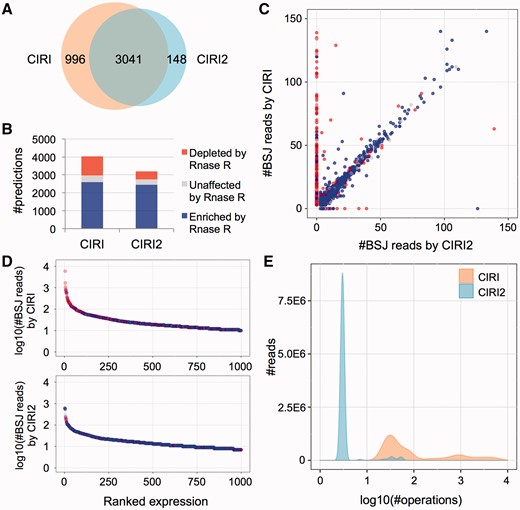
Performance comparison between CIRI and CIRI2. (A) Overlap of circRNA predictions by CIRI2 and CIRI on Hs68 data sets. (B) RNase R resistance of the CIRI and CIRI2 predictions for Hs68 data sets. (C) RNase R resistance and BSJ read count detected for each prediction by CIRI and CIRI2. Only predictions with BSJ read counts ≤150 are shown. (D) RNase R resistance of top 1000 most abundant predictions. Blue nodes indicate the circRNAs enriched by RNase R treatment, whereas red nodes indicate the circRNAs depleted by RNase R treatment. (E) Comparison of computing operation counts of CIRI and CIRI2 for the Hs68 data set (SRR444975).
To further explore the efficiency on filtering false positives of the adapted MLE in CIRI2 and the dynamic programming alignment in CIRI, we scrutinized the mapping details of BSJ reads for abundant candidates only predicted by CIRI. We found that these false predictions could be mainly attributed to repetitive sequences across human chromosomes or unplaced contigs in the human genome assembly. For example, a candidate circRNA (chr8: 70 602 323|70 602 497) had 14 BSJ reads according to CIRI prediction, but our manual curation showed that most of these BSJ reads indeed could be aligned to repetitive regions of an unplaced contig (Un_gl000220) at a higher identity compared with chr8. Because one of the 14 reads was wrongly mapped to chr8, the dynamic programming alignment implemented in CIRI treated other 13 reads as ‘unbalanced junction reads’ and, thus, lead to false prediction. In CIRI2, however, the determination of a BSJ read was based on the individual MLE according to multiple seed matching rather than its sequence similarity with other BSJ reads, and, therefore, such false positives can be largely avoided. Indeed, the majority of abundant predictions (74.2% of 416 with more than five BSJ reads) exclusively detected by CIRI could be aligned to unplaced contigs at a high sequence identity (>90%) or contained a high percentage of unreliable ‘unbalanced BSJ reads’ (>75%) attributed to the dynamic programming alignment in CIRI. In contrast, such percentage was much lower in the predictions reported by both CIRI and CIRI2 as well as those exclusively detected by CIRI2 (3.5% of 1244 and 3.3% of 30, respectively).
We further counted the computing operations of CIRI and CIRI2 when using them to process the above data set. As shown in Figure 2E, sequencing reads required a significant amount of computing operations (10–10 000 per read) in CIRI, whereas MLE based on multiple seed matching in CIRI2 could make a decision in merely several operations for the vast majority of sequencing reads to determine whether they were from BSJs. As the operation times largely reflects time complexity and determines the running time of the algorithm, the above evaluation indicated that the optimization of CIRI2 has much higher computation efficiency compared with CIRI.
Comparison on sensitivity and reliability
We next applied eight widely used algorithms including CIRCexplorer [11], circRNA_finder [1], DCC [25], find_circ [8], KNIFE [17], MapSplice [16], segemehl [18] and UROBORUS [26] to the same data sets and compared their performance with CIRI2. Using the criteria described above, we found that all of these existing algorithms showed some biased performance on detecting true positives and filtering false positives (Figure 3A). For example, although segemehl achieved the highest sensitivity, it also resulted in a large amount of false positives. On the other hand, MapSplice showed excellent ability in controlling the FDR, but it could only identify about two-thirds of the true positives detected by segemehl. In contrast, CIRI2 had much more balanced performance. It exhibited similar low FDR with MapSplice and CIRCexplorer and simultaneously achieved comparable sensitivity with segemehl. We further tested the above tools on the 150 bp HEK293 data set. Similarly, CIRI2 showed both high sensitivity and low FDR, whereas all other algorithms were inferior to CIRI2 in at least one of the two metrics (Figure 3B), regardless of the expression levels of circRNAs (Supplementary Figure S4).
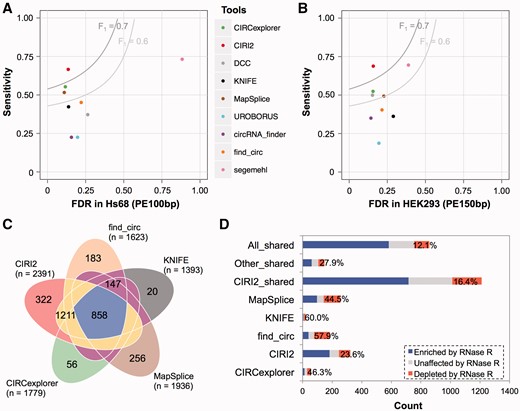
Comparison of CIRI2 and other eight circRNA detection methods. (A) Sensitivity and FDR of CIRI2 and other eight methods for the Hs68 data sets. (B) Sensitivity and FDR of CIRI2 and other eight methods for the HEK293 data sets. (C) Overlap of circRNA predictions by CIRI2 and other four frequently used methods on the HEK293 data sets. (D) RNase R resistance of the exotic and shared predictions by CIRI2 and the other four frequently used methods.
To better understand the overall performance of these tools, we defined an evaluation metric, F1 score, which equally favors increase of sensitivity and decrease of FDR. As shown in Supplementary Figure S5, CIRI2 had the highest F1 scores in both data sets (Hs68: 0.752 and HEK293: 0.757) in all of the nine algorithms. When applying different thresholds of RNase R enrichment to determine true positives, CIRI2 outperformed all other algorithms in all of the cases (Supplementary Figure S6), which demonstrates the stable performance of CIRI2 on detecting circRNAs with different resistance to RNase R. We next selected other four algorithms frequently used in previous studies (CIRCexplorer, MapSplice, find_circ and KNIFE), and compared their predicted circRNAs with those detected by CIRI2. As shown in Figure 3C, all five algorithms shared 858 predictions in the data set of HEK293, whereas 837 were exclusively detected by only one algorithm, which were termed as exotic circRNAs in the previous study [21]. As exotic circRNAs could effectively reflect both reliability and indispensability of an algorithm, we scrutinized exotic circRNAs of all five algorithms. As shown in Figure 3D, exotic circRNAs of CIRI2 contained the lowest percentage (23.6%) of false positives, while the other four tools were enriched with false positives in their exotic circRNAs (44.5–60%). Meanwhile, CIRI2 also exclusively detected the largest number of true positives (182 of 322 exotic circRNAs), which were even more than the sum of all other four tools (157 of 515 exotic circRNAs in total). Besides the exotic circRNAs and predictions shared by all five tools, there were 1211 predictions shared by CIRI2 and at least one of the other tools, which were significantly >147 predictions exclusively shared by other tools (Wilcoxon rank test, P < 0.01), indicating that circRNAs detected by CIRI2 were more likely to be supported by other independent tools. Moreover, the CIRI2-related group contained much more true positives (716 versus 61) and lower percentage of false positives (16.4 versus 27.9%).
Taken together, the above performance evaluations demonstrated that CIRI2 outperformed all currently available tools on detecting circRNAs, and it could not be replaced by any of the other eight algorithms or their combinations.
Comparison on duration, RAM usage and dependencies
We next asked whether the good performance of CIRI2 is based on long duration or high memory usage. The largest data set from Hs68 and the RNase R-treated data set from HEK293 were used to benchmark the computation resources required by all 10 tools including CIRI and CIRI2. It should be noted that all of these tools are dependent on specific mappers. Therefore, we compared computation resources required by the whole pipeline recommended by developer instead of the circRNA detection step itself. We found that most of the pipelines with better performances tended to need longer running time. For example, MapSplice and CIRCexplorer pipeline ran for >60 h on the 41.3 Gb data set of Hs68 when 10 threads were used (Figure 4A). However, CIRI2 was exceptional—its whole pipeline including generating SAM and CIRI2 analysis only spent about 8 h on the same data set, which ranked the second fastest tool (Figure 4A and Table 1). The running time of CIRI2 step could be further shortened through multithreading. For example, it took only 1.5 h when 10 threads were used (Figure 4C).
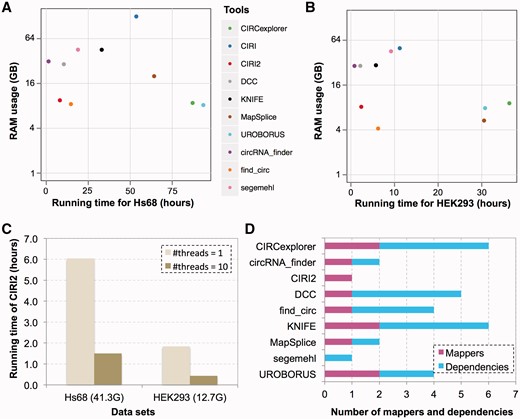
Duration, RAM usage and dependencies of CIRI2 and other detection methods. (A) Duration and RAM usage of 10 tools for the Hs68 data set (SRR444975). Single thread was used for CIRI2. (B) Duration and RAM usage of 10 algorithms for the HEK293 data set (SRR3479244). Single thread was used for CIRI2. (C) Running time of CIRI2 for both data sets when single thread or 10 threads were used. (D) Number of mappers and dependencies of CIRI2 and other eight methods.
Overview of CIRI2 and other eight circRNA detection tools
| Tool | De novo | Mapper | Dependencies | Language | Multithreada | Version | Sensitivity/FDR/F1 score | Duration (hours)/RAM (G) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Hs68 (PE100 bp) | HEK293 (PE150 bp) | Hs68 (41.3 Gb) | HEK293 (12.7 Gb) | |||||||
| CIRCexplorer | No | Bowtie | Tophat bedtools | Python | No | 1.1.7 | 0.553/0.117/0.680 | 0.524/0.156/0.646 | 87.13/8.75 | 36.32/9.12 |
| Bowtie2 | Pysam docopt | |||||||||
| circRNA_finder | Yes | STAR | Samtools | Perl | No | N/A | 0.225/0.157/0.356 | 0.350/0.143/0.497 | 1.34/31.65 | 0.82/28.94 |
| CIRI2 | Yes | BWA | None | Perl | Yes | 2.0.2 | 0.666/0.136/0.752 | 0.688/0.158/0.757 | 8.08/9.50 | 2.38/8.17 |
| DCC | No | STAR | Pysam pandas | Python | Yes | 0.4.4 | 0.372/0.264/0.494 | 0.499/0.153/0.628 | 10.44/29.02 | 2.16/28.99 |
| Numpy HTSeq | ||||||||||
| find_circ | Yes | Bowtie2 | Pysam samtools | Python | No | 1.2 | 0.451/0.221/0.571 | 0.404/0.215/0.533 | 14.66/8.45 | 6.24/4.18 |
| Bedtools | ||||||||||
| KNIFE | No | Bowtie | Samtools numpy | Perl R | No | 1.4 | 0.423/0.138/0.567 | 0.362/0.289/0.480 | 33.00/45.36 | 5.74/29.52 |
| Bowtie2 | scipy data.table | Python | ||||||||
| MapSplice | No | Bowtie | Samtools | Python C ++ | Yes | 2.2.0 | 0.516/0.110/0.654 | 0.493/0.227/0.602 | 64.23/20.03 | 30.53/5.34 |
| segemehl | Yes | Per se | Samtools | C | Yes | 0.2.0 | 0.732/0.883/0.201 | 0.695/0.389/0.650 | 18.86/45.49 | 9.2/45.46 |
| UROBORUS | No | Bowtie | Tophat samtools | Perl | No | 0.1.3 | 0.225/0.197/0.352 | 0.187/0.194/0.304 | 93.52/8.20 | 30.76/7.85 |
| Bowtie2 | ||||||||||
| Tool | De novo | Mapper | Dependencies | Language | Multithreada | Version | Sensitivity/FDR/F1 score | Duration (hours)/RAM (G) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Hs68 (PE100 bp) | HEK293 (PE150 bp) | Hs68 (41.3 Gb) | HEK293 (12.7 Gb) | |||||||
| CIRCexplorer | No | Bowtie | Tophat bedtools | Python | No | 1.1.7 | 0.553/0.117/0.680 | 0.524/0.156/0.646 | 87.13/8.75 | 36.32/9.12 |
| Bowtie2 | Pysam docopt | |||||||||
| circRNA_finder | Yes | STAR | Samtools | Perl | No | N/A | 0.225/0.157/0.356 | 0.350/0.143/0.497 | 1.34/31.65 | 0.82/28.94 |
| CIRI2 | Yes | BWA | None | Perl | Yes | 2.0.2 | 0.666/0.136/0.752 | 0.688/0.158/0.757 | 8.08/9.50 | 2.38/8.17 |
| DCC | No | STAR | Pysam pandas | Python | Yes | 0.4.4 | 0.372/0.264/0.494 | 0.499/0.153/0.628 | 10.44/29.02 | 2.16/28.99 |
| Numpy HTSeq | ||||||||||
| find_circ | Yes | Bowtie2 | Pysam samtools | Python | No | 1.2 | 0.451/0.221/0.571 | 0.404/0.215/0.533 | 14.66/8.45 | 6.24/4.18 |
| Bedtools | ||||||||||
| KNIFE | No | Bowtie | Samtools numpy | Perl R | No | 1.4 | 0.423/0.138/0.567 | 0.362/0.289/0.480 | 33.00/45.36 | 5.74/29.52 |
| Bowtie2 | scipy data.table | Python | ||||||||
| MapSplice | No | Bowtie | Samtools | Python C ++ | Yes | 2.2.0 | 0.516/0.110/0.654 | 0.493/0.227/0.602 | 64.23/20.03 | 30.53/5.34 |
| segemehl | Yes | Per se | Samtools | C | Yes | 0.2.0 | 0.732/0.883/0.201 | 0.695/0.389/0.650 | 18.86/45.49 | 9.2/45.46 |
| UROBORUS | No | Bowtie | Tophat samtools | Perl | No | 0.1.3 | 0.225/0.197/0.352 | 0.187/0.194/0.304 | 93.52/8.20 | 30.76/7.85 |
| Bowtie2 | ||||||||||
Note. For each evaluation, values of the best two tools are shown in bold.
aThis column shows whether the corresponding tool itself is multithreaded, rather than whether it invokes or depends on some other multithreaded tools.
Overview of CIRI2 and other eight circRNA detection tools
| Tool | De novo | Mapper | Dependencies | Language | Multithreada | Version | Sensitivity/FDR/F1 score | Duration (hours)/RAM (G) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Hs68 (PE100 bp) | HEK293 (PE150 bp) | Hs68 (41.3 Gb) | HEK293 (12.7 Gb) | |||||||
| CIRCexplorer | No | Bowtie | Tophat bedtools | Python | No | 1.1.7 | 0.553/0.117/0.680 | 0.524/0.156/0.646 | 87.13/8.75 | 36.32/9.12 |
| Bowtie2 | Pysam docopt | |||||||||
| circRNA_finder | Yes | STAR | Samtools | Perl | No | N/A | 0.225/0.157/0.356 | 0.350/0.143/0.497 | 1.34/31.65 | 0.82/28.94 |
| CIRI2 | Yes | BWA | None | Perl | Yes | 2.0.2 | 0.666/0.136/0.752 | 0.688/0.158/0.757 | 8.08/9.50 | 2.38/8.17 |
| DCC | No | STAR | Pysam pandas | Python | Yes | 0.4.4 | 0.372/0.264/0.494 | 0.499/0.153/0.628 | 10.44/29.02 | 2.16/28.99 |
| Numpy HTSeq | ||||||||||
| find_circ | Yes | Bowtie2 | Pysam samtools | Python | No | 1.2 | 0.451/0.221/0.571 | 0.404/0.215/0.533 | 14.66/8.45 | 6.24/4.18 |
| Bedtools | ||||||||||
| KNIFE | No | Bowtie | Samtools numpy | Perl R | No | 1.4 | 0.423/0.138/0.567 | 0.362/0.289/0.480 | 33.00/45.36 | 5.74/29.52 |
| Bowtie2 | scipy data.table | Python | ||||||||
| MapSplice | No | Bowtie | Samtools | Python C ++ | Yes | 2.2.0 | 0.516/0.110/0.654 | 0.493/0.227/0.602 | 64.23/20.03 | 30.53/5.34 |
| segemehl | Yes | Per se | Samtools | C | Yes | 0.2.0 | 0.732/0.883/0.201 | 0.695/0.389/0.650 | 18.86/45.49 | 9.2/45.46 |
| UROBORUS | No | Bowtie | Tophat samtools | Perl | No | 0.1.3 | 0.225/0.197/0.352 | 0.187/0.194/0.304 | 93.52/8.20 | 30.76/7.85 |
| Bowtie2 | ||||||||||
| Tool | De novo | Mapper | Dependencies | Language | Multithreada | Version | Sensitivity/FDR/F1 score | Duration (hours)/RAM (G) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Hs68 (PE100 bp) | HEK293 (PE150 bp) | Hs68 (41.3 Gb) | HEK293 (12.7 Gb) | |||||||
| CIRCexplorer | No | Bowtie | Tophat bedtools | Python | No | 1.1.7 | 0.553/0.117/0.680 | 0.524/0.156/0.646 | 87.13/8.75 | 36.32/9.12 |
| Bowtie2 | Pysam docopt | |||||||||
| circRNA_finder | Yes | STAR | Samtools | Perl | No | N/A | 0.225/0.157/0.356 | 0.350/0.143/0.497 | 1.34/31.65 | 0.82/28.94 |
| CIRI2 | Yes | BWA | None | Perl | Yes | 2.0.2 | 0.666/0.136/0.752 | 0.688/0.158/0.757 | 8.08/9.50 | 2.38/8.17 |
| DCC | No | STAR | Pysam pandas | Python | Yes | 0.4.4 | 0.372/0.264/0.494 | 0.499/0.153/0.628 | 10.44/29.02 | 2.16/28.99 |
| Numpy HTSeq | ||||||||||
| find_circ | Yes | Bowtie2 | Pysam samtools | Python | No | 1.2 | 0.451/0.221/0.571 | 0.404/0.215/0.533 | 14.66/8.45 | 6.24/4.18 |
| Bedtools | ||||||||||
| KNIFE | No | Bowtie | Samtools numpy | Perl R | No | 1.4 | 0.423/0.138/0.567 | 0.362/0.289/0.480 | 33.00/45.36 | 5.74/29.52 |
| Bowtie2 | scipy data.table | Python | ||||||||
| MapSplice | No | Bowtie | Samtools | Python C ++ | Yes | 2.2.0 | 0.516/0.110/0.654 | 0.493/0.227/0.602 | 64.23/20.03 | 30.53/5.34 |
| segemehl | Yes | Per se | Samtools | C | Yes | 0.2.0 | 0.732/0.883/0.201 | 0.695/0.389/0.650 | 18.86/45.49 | 9.2/45.46 |
| UROBORUS | No | Bowtie | Tophat samtools | Perl | No | 0.1.3 | 0.225/0.197/0.352 | 0.187/0.194/0.304 | 93.52/8.20 | 30.76/7.85 |
| Bowtie2 | ||||||||||
Note. For each evaluation, values of the best two tools are shown in bold.
aThis column shows whether the corresponding tool itself is multithreaded, rather than whether it invokes or depends on some other multithreaded tools.
RAM usage directly determines whether an algorithm can be conveniently run on any available server with limited RAM. We monitored RAM usage of all 10 pipelines, and found that they could be divided into two groups according to their RAM usage. Segemehl, circRNA_finder, UROBORUS, DCC, CIRCexplorer and CIRI2 pipelines showed relatively stable RAM usage regardless of data size, whereas the other four pipelines needed much more RAM (1.5- to 3.8-fold) for the 41.3 Gb Hs68 data set than the 12.7 Gb HEK293 data set (Figure 4A and B). As to actual RAM usage, most of the tools needed >20 Gb RAM for at least one of the data sets, and may call for moderate or high-performance server. In contrast, find_circ, UROBORUS, CIRI2 and CIRCexplorer pipelines used <10 Gb RAM for both data sets (Figure 4A and B), and therefore were able to run on personal computer or small server.
To further compare usability of these detection methods, we also summarized dependencies, mappers and steps required by them. As shown in Figure 4D and Table 1, none of the other eight methods could independently complete detection of circRNAs, and some of them even depended on three to four additional tools or non-default packages for installation and running. In contrast, CIRI2 was the only standalone tool of all. As to mappers, segemehl could complete mapping of sequencing reads all by itself, whereas other tools needed to invoke or analyze mapping output of one (circRNA_finder, CIRI2, DCC, find_circ and MapSplice) or two (CIRCexplorer, KNIFE and UROBORUS) additional tools. We also summarized the steps of the corresponding pipelines, and found these tools needed one to four steps to complete their detection (Supplementary Figure S7 and Supplementary Table S1).
Discussion
Detection of circRNA loci is usually an initial and important step for in-depth study of circRNAs, such as binding site prediction, internal structure identification, expression analysis and function exploration. Although several computational methods have been developed for circRNA detection, different studies that evaluated and compared these methods often drew contradictory conclusions. Such inconsistency can be attributed to the lack of a highly confident database and the distinct basis of the criteria adopted by the studies. Tests based on simulated data were commonly used in such evaluation, but it is difficult to evaluate whether the related simulation methods themselves were reasonable. Moreover, simulated data can hardly mimic complexities of eukaryotic transcription and splicing, and thus are not ideal choice for circRNA detection evaluation by now. In contrast, real data from RNase R-treated samples can provide relatively objective assessments. BSJ read count enrichment evaluation in these samples was first used to confirm the reliability of predictions in a pipeline [27], and then it was adapted to estimate FDR of five detection methods [21]. However, without estimating sensitivity of methods, comparison of the detection methods, especially on the basis of single threshold of RNase R enrichment, remains to be partial. Here, we adopted the same approach for FDR estimation, and simultaneously introduced sensitivity based on all RNase R-resistant predictions of the methods to be compared. The F1 scores, calculated based on the estimated FDR and sensitivity by applying multiple thresholds of RNase R enrichment, facilitate a direct comparison of circRNA detection methods and represents an impartial metric that could be widely used in future.
A well-designed circRNA detection method should have good performances on sensitivity, FDR, duration and RAM usage, but with few dependencies for its implementation. The metric of sensitivity evaluates the percentage of circRNAs that can be detected by a method and the possibility of the method to uncover unknown circRNAs or functions. FDR reflects the reliability of each prediction as well as whether it could be used for experimental validation and further analyses. RAM usage and dependencies decide the availability and feasibility of a tool on limited computation resource, and duration largely concerns how long a method could generate results available for further study. It should be mentioned that our previous study [21] demonstrated, because of the relatively low abundance of most circRNAs, in-depth studies on circRNAs such as alternative splicing analysis needed much more data than previously thought, which will largely challenge detection tools on their duration and RAM usage.
Here, we used an adapted MLE to identify BSJ reads. Compared with simply analyzing limited mapping records or similarity with other reads, such MLE considers both possible regions of undetermined segment in a potential BSJ read, and thus is highly effective in filtering false positives derived from erroneous mapping or repetitive sequences in the genome. Also, as the underlying multiple seed matching can tolerate sequencing errors or interrupted eukaryotic transcription but in the same time substantially reduce computing operations, it facilitates the efficient and prompt implementation of the above MLE in de novo circRNA detection. Through extensive performance evaluations, CIRI2 was demonstrated to have the best average F1 score in the test data sets, and need shorter running time and lower RAM usage than most of other algorithms, with no requirement of dependencies. Because of extensive attention and limited knowledge on circRNAs, we believe that CIRI2 provides an efficient and unbiased circRNA detection approach for future circRNA studies.
An adapted MLE model with dramatically reduced FDR and running time.
Establish objective assessment criteria based on real data sets from RNase R-treated samples.
CIRI2 shows the best performance among 10 popular circRNA detection tools.
Funding
The National Natural Science Foundation of China (grant numbers 91640117 and 91531306), the National Key R&D Program (grant number 2016YFC1200804) and the Strategic Priority Research Program of the Chinese Academy of Sciences (grant number XDB13000000).
Yuan Gao is a PhD student at University of Chinese Academy of Sciences.
Jinyang Zhang is a master student at University of Chinese Academy of Sciences.
Fangqing Zhao is a professor at Beijing Institutes of Life Science, Chinese Academy of Sciences.

{kind=link}
{kind=link}
{kind=link}
{kind=link}